Eukaryotic 18S rRNA Sequencing
Service Overview
What Is Eukaryotic 18S rRNA?
The 18S rRNA gene encodes for the small subunit rRNA in eukaryotic organisms, reflecting the species variation amongst eukaryotic samples. Similar to bacterial diversity analysis, three types of ribosomal RNA (rRNA) exist within eukaryotic microorganisms: 5.8S rRNA, 18S rRNA, and 28S rRNA. The 18S rRNA gene, which encodes for the small subunit in eukaryotic ribosomes, encompasses both conserved and variable regions (V1-V9, lacking the V6 region). The conserved regions reflect phylogenetic relationships between species, while the variable regions manifest interspecies variability, lending utility to species-level and above classification parameters. Notably, the V4 region frequently serves as the analytical fragment in scholarly literature.
Our eukaryotic 18S rRNA sequencing applies both Next-Generation Sequencing (NGS) and long-read sequencing platforms to identify specific 18S ribosomal RNA (rRNA) genes in eukaryotes and yeasts. This service aids in uncovering the structure of microbial communities, the evolutionary relationships of eukaryotic organisms, and the correlations between eukaryotes and their environment.
Our Advantages:
-
Three sequencing platforms: Illumina PE250/300, Nanopore platforms, and PacBio platforms.
- Long-read sequencing can deliver full-length 18S rRNA sequences without the need for assembly.
- Strict data quality control: a rigorous quality control system ensures accuracy.
- High-quality data assurance, diverse analytical content.
- Clear workflow, minimized sample and time waste, short turnaround time.
- Decades of experience in microbial genomics services.
Application of Eukaryotic 18S rRNA sequencing
The 18S rRNA sequences of eukaryotes serve as common markers for molecular systematics and evolutionary research. As a constituent component of the small subunit ribosome, it possesses highly conserved sequences and structural features, rendering it one of the crucial tools for studying phylogeny and taxonomy of eukaryotic organisms. The sequencing of the 18S rRNA gene is typically employed for the identification, classification, and quantification of microbes within complex biological mixtures--for instance, samples derived from environmental and gastrointestinal sources. Phylogenetic trees can be constructed utilizing various eukaryotic 18S rRNA gene sequences to scrutinize the genetic diversity and evolutionary relationships of eukaryotes. Given the pivotal roles that eukaryotic microbes undertake as decomposers, predators, producers, and parasites in the majority of ecological niches, 18S rRNA sequencing proves to be a potent tool enabling an enhanced understanding of ecosystems through the analysis of eukaryotic organism classification, evolution, ecology, and diversity. Moreover, 18S rRNA sequencing can also be applied in the identification of eukaryotic organisms associated with human and animal diseases.
Difference Between ITS and 18S sequencing
Diverging in their target sequences, Internal Transcribed Spacer (ITS) sequences are situated in the transcriptional interspace regions of ribosomal RNA (rRNA) gene constituents, typically encompassing both ITS1 and ITS2 sub-regions. In contrast, 18S rRNA, a smaller ribosomal fraction, is a component of the minuscule ribosomal RNA gene subset within the corpus of rRNA genes. Sequence lengths further differentiate these sequences: ITS sequences are habitually shorter with high sequence variability, whereas 18S rRNA sequences are noticeably longer and relatively conserved.
As such, their applications in scientific domains also diverge. ITS sequences find common use in species identification, intra- and inter-species phylogenetic analyses, biological diversity surveys, and microbial community structure analyses, among others. Yet, 18S rRNA sequences also find usage in the same domains like phylogenetic analysis, biodiversity surveys, and microbial community structural analysis of environmental samples, although their relatively conservative nature makes them particularly advantageous for studies concerning higher-level taxonomic units.
Difference Between 16s rRNA and 18S rRNA
Cell Types:
16S rRNA: is found in the ribosomes of prokaryotic organisms, such as bacteria and archaea.
18S rRNA: is found in the ribosomes of eukaryotes, including plants, animals, fungi, and others.
Functions:
16S rRNA: It participates in the assembly of prokaryotic ribosomes, playing a crucial part in supporting ribosome formation and protein synthesis.
18S rRNA: Involved in the construction of eukaryotic ribosomes, it also serves a pivotal role in ribosome formation and protein synthesis.
Sequence Length and Structure:
16S rRNA: Its length generally falls between about 1,500 to 1,600 nucleotides, featuring nine highly-conserved regions and nine variable regions. The structural and sequenced variation in these regions can be used to differentiate between various bacteria and archaea.
18S rRNA: Its length usually falls between about 1,800 to 2,000 nucleotides. It also has highly-conserved and variable regions. However, dynamically different from 16S rRNA, fluctuations in 18S rRNA sequence and structure are adopted to discern different eukaryotic organisms, such as plants, animals, fungi, and others.
Applications:
16S rRNA: Frequently used in microbiological research, such as in bacterial taxonomy, community structure analysis, and environmental microbial studies.
18S rRNA: Commonly used for phylogenetic and taxonomic studies of eukaryotes and in ecological investigation, such as biodiversity assessments and population genetics research.
Service Specifications
Introduction to Our Eukaryotic 18S rRNA sequencing Service

The 18S rRNA genes form a conservative component of eukaryotic genomes. These include both conservative and variable regions (V1 to V9, excluding V6 which is relatively conservative), reflecting the interspecies differences among eukaryotes such as fungi. The 18S rRNA sequencing continues to be the gold standard for eukaryotic molecular phylogeny. Our integrated 18S rRNA sequencing platform combines advanced sequencing technology and quantitative PCR (q-PCR) for accurate analysis of the 18S rRNA gene. This platform allows short read length 18S rRNA sequencing via Next-Generation Sequencing (NGS - Illumina PE250/300), or full-length 18S rRNA sequencing through PacBio SMRT sequencing or Nanopore sequencing. The long-read sequencing technology overcomes the limitations of read length and delivers more precise species-level identification. Quantitative PCR can be utilized for the quantification of microbial species and validation of sequencing data. Moreover, we offer fungal ITS sequencing for the diversity analysis of fungi.
Bioinformatics Analysis
Our bioinformatics analysis includes five parts: community composition analysis, diversity analysis, evolutionary analysis, functional analysis, and other analyses. We are flexible to your needs.
| Bioinformatics analysis |
Details |
| Community composition analysis |
OUT clustering, Species annotation, heatmap, taxonomic tree, etc. |
| Diversity analysis |
α diversity, β diversity, meta-analysis |
| Evolutionary analysis |
Construction of phylogenetic trees, estimation of genetic distance |
| Functional analysis |
PICRUSt, FAPROTAX, Tax4fun, FunGuild, LDA, LEfSe |
| Other analyses |
VPA analysis, RDA/CCA, network analysis, etc. |
Eukaryotic 18S rRNA sequencing workflow
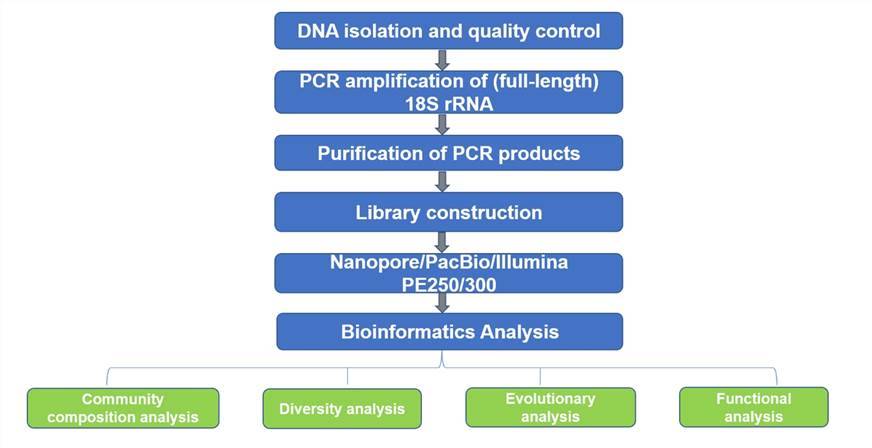
Deliverables
- Raw sequencing data (FASTQ)
- Trimmed and stitched sequences (FASTA)
- Quality-control dashboard
- Statistic data
- Your designated bioinformatics reports
FAQs
Eukaryotic 18S rRNA Sequencing FAQs
- Why 18S rRNA is used for eukaryotes?
- What is 18S rRNA sequencing for?
- What is the function of 18S rRNA?
Demo
Demo
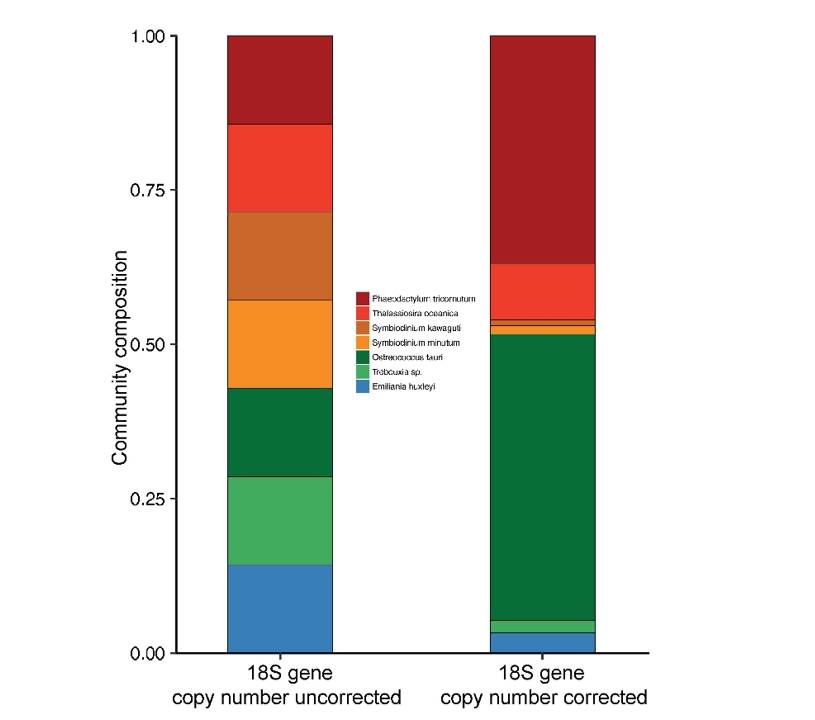 Estimation of 18S gene copy number in marine eukaryotic plankton using a next-generation sequencing approach (Gong et al., 2019)
Estimation of 18S gene copy number in marine eukaryotic plankton using a next-generation sequencing approach (Gong et al., 2019)
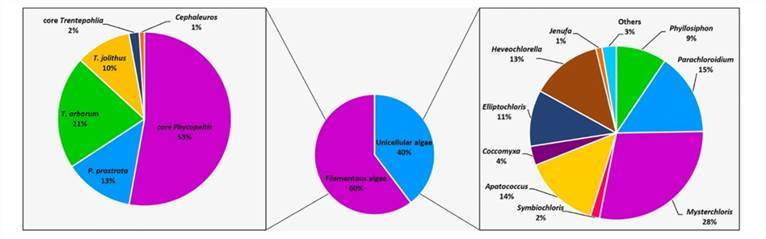 Molecular characterization of eukaryotic algal communities in the tropical phyllosphere based on real-time sequencing of the 18S rDNA gene. (Zhu et al., 2018)
Molecular characterization of eukaryotic algal communities in the tropical phyllosphere based on real-time sequencing of the 18S rDNA gene. (Zhu et al., 2018)
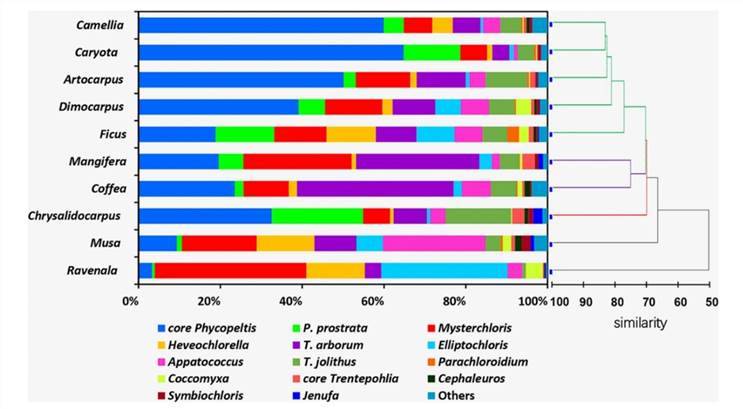 Molecular characterization of eukaryotic algal communities in the tropical phyllosphere based on real-time sequencing of the 18S rDNA gene. (Zhu et al., 2018)
Molecular characterization of eukaryotic algal communities in the tropical phyllosphere based on real-time sequencing of the 18S rDNA gene. (Zhu et al., 2018)
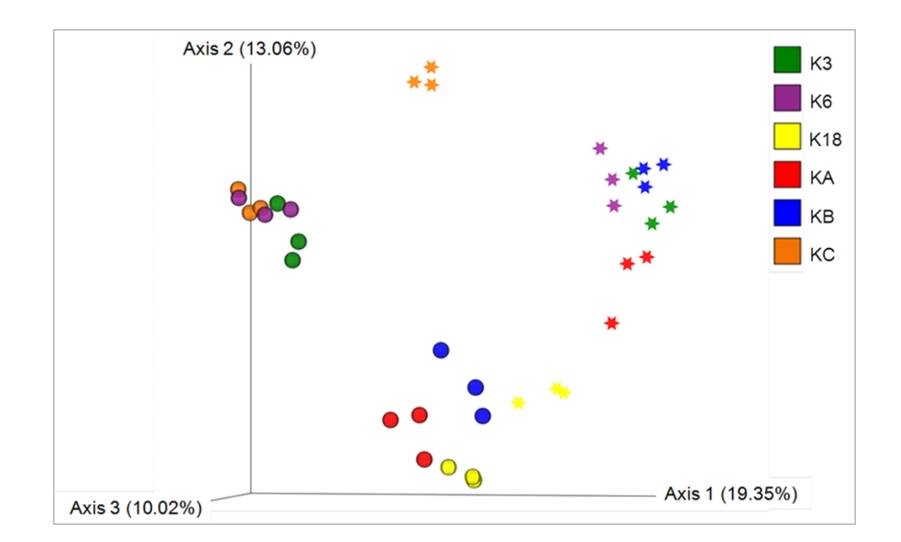 Distribution and diversity of eukaryotic microalgae in Kuwait waters assessed using 18S rRNA gene sequencing. (Kumar et al., 2021)
Distribution and diversity of eukaryotic microalgae in Kuwait waters assessed using 18S rRNA gene sequencing. (Kumar et al., 2021)
Case Study
18S rRNA gene sequencing reveals significant influence of anthropogenic effects on microeukaryote diversity and composition along a river-to-estuary gradient ecosystem
Journal: Science of the total environment
Impact factor: 9.8
Published: 1 December 2019
Backgrounds
The pivotal role of microeukaryotes in the structure and function of aquatic ecosystems is well acknowledged; however, our understanding of the relative significance of biogeographical drivers in riverine microeukaryotic communities, especially the impact of anthropogenic inputs such as wastewater discharge and the use of pesticides and fertilizers on microeukaryotic taxonomic and functional diversity, is still in its infancy. In this study, 18S rRNA sequencing technology was employed to probe the assembly of microeukaryotic communities in samples from a medium-sized river in northern China: the Xiaoqing River.
Methods
Sample preparation:
-
Eukaryotic plankton collection
- DNA extraction
- Sequencing:
- PCR amplicon libraries
- 454/Roche
- 18S rRNA sequencing
Data analysis:
-
Taxonomic diversity
- Observed richness
- Shannon–Wiener diversity
- Pielou's evenness
- Rarefaction curves
Results
1. Community diversity
The findings demonstrate that the total operational taxonomical units (OTUs) for X1, X2, X3, X4, and X5 were 475, 396, 429, 450, and 413 respectively, presenting a comparable level of OTUs across these locations. However, location X0 exhibited a significantly higher level of total OTUs at 705. This implies a greater level of species richness in location X0 compared to the others. Additionally, it was observed that both upstream and downstream locations of the Xiaoqing River presented high biodiversity, while midstream locations showcased less due to anthropogenic disturbances.
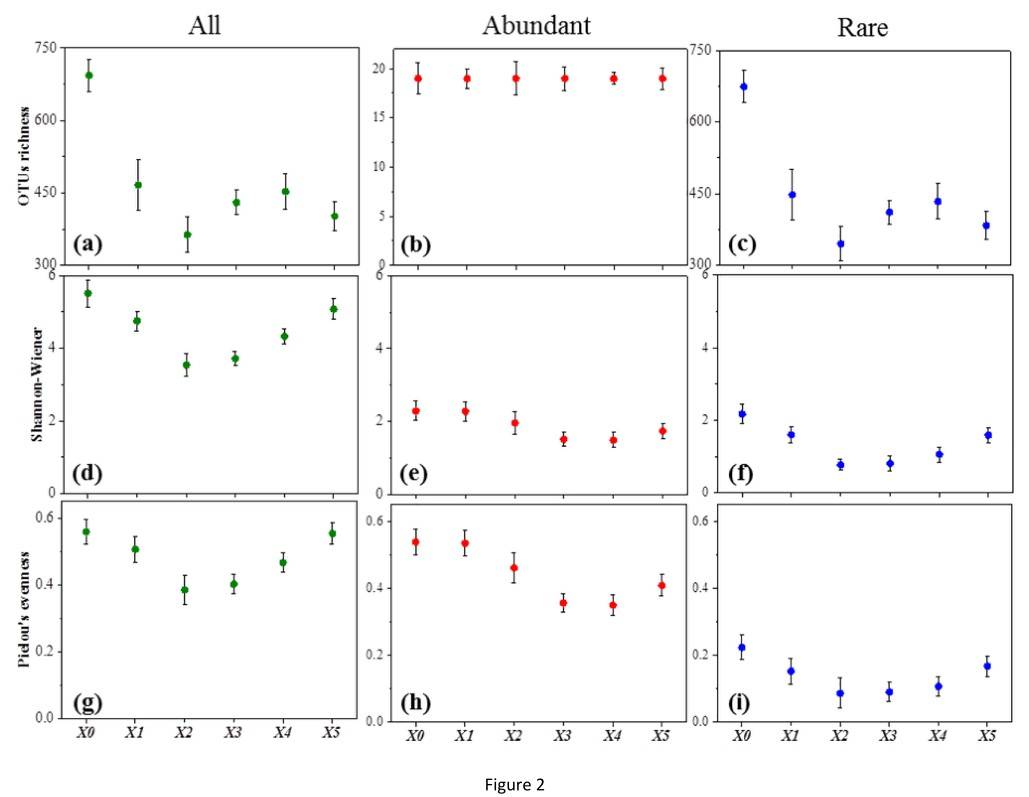 Figure 1. Variation in alpha-diversity with different sites.
Figure 1. Variation in alpha-diversity with different sites.
2. Overall eukaryotic community structure
The species composition of each sample in the study reflected the dominant taxa of its origin. Pigmented taxa, primarily Ochrophyta, Chloroplastida, and Cryptomonadales, constituted between 35.2-81.9% of the compositions. In contrast, the Xiaoqing River saw prevalent non-pigmented taxa such as Cercozoa, Ciliophora, Rotifera, and Arthropoda, essentially representing the eukaryotic plankton. Moreover, rarely seen non-pigmented taxa were dominant in non-pigmented communities.
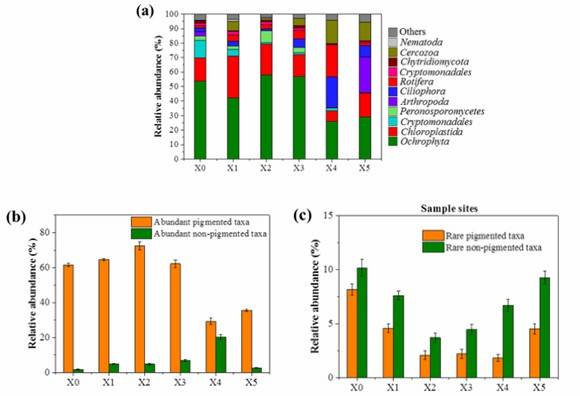 Figure 2. (a) Relative abundance of the predominant phyla for each site (with average sequence percentage above 1.0%); Relative abundance of the abundant taxa (b) and rare taxa (c) for each site.
Figure 2. (a) Relative abundance of the predominant phyla for each site (with average sequence percentage above 1.0%); Relative abundance of the abundant taxa (b) and rare taxa (c) for each site.
3. Environmental factors associated with patterns of eukaryotic community structure
The results of this study present that the disparity in community composition between any two locations housing micro-eukaryotic life forms escalates as the Euclidean distance of environmental variables increases. Spearman's correlation demonstrated a significant positive correlation between the Bray-Curtis community dissimilarity of micro-eukaryotic communities and Euclidean distance of environmental variables, with an overall correlation coefficient of 0.591 (p <0.05). The impact of environmental variables on pigment taxon showed no statistical significance (p=0.097); on the other hand, the influence of environmental variables on non-pigment taxa manifested statistical significance (p <0.01). Redundancy analysis (RDA) identifies nutrients (TN, TP), dissolved oxygen (DO), turbidity, Cl− and salinity as strong factors affecting the composition and distribution of all taxa and rare taxa in the micro-eukaryotic communities.
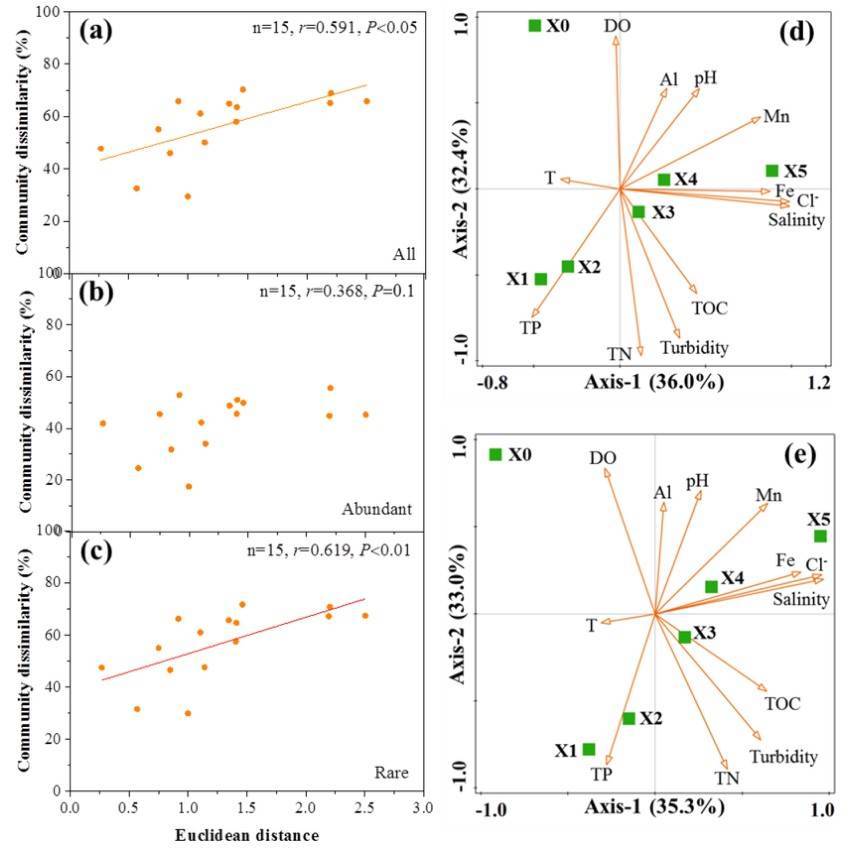 Figure 3. Relationships between microeukaryotic communities and environmental factors.
Figure 3. Relationships between microeukaryotic communities and environmental factors.
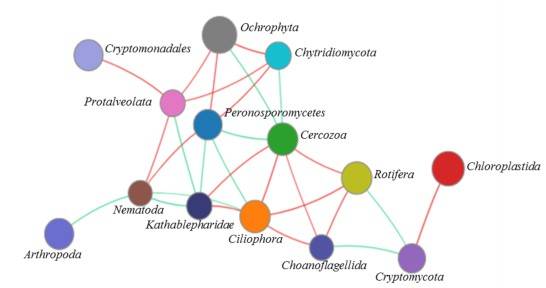 Figure 4. Network analysis of the microeukaryotic community at the phylum level.
Figure 4. Network analysis of the microeukaryotic community at the phylum level.
Conclusion
The observed findings declare that in ecologically significant gradiented water systems, localized pollutants can largely determine the community structure of picoeukaryotes. Their compositional variations escalate with the Euclidean distance of environmental variables. The variations documented in the diversity of picoeukaryotes are predominantly determined by fluctuations in nutrient levels, dissolved oxygen, turbidity, and salinity, exerting significant impact on rare subcommunities. Moreover, zooplankton is primarily composed of rare taxa, whilst phytoplankton is predominantly replete with abundant taxa. These findings reinforce the dynamic nature of riverine ecosystems and underscore the significant role human activities play in shaping the diversity of riverine picoeukaryotes.
References
- Wang Y, Tian R M, Gao Z M, et al. Optimal eukaryotic 18S and universal 16S/18S ribosomal RNA primers and their application in a study of symbiosis. PloS one, 2014, 9(3): e90053.
- Gong W, Marchetti A. Estimation of 18S gene copy number in marine eukaryotic plankton using a next-generation sequencing approach. Frontiers in marine Science, 2019, 6: 219.
- Zhu H, Li S, Hu Z, et al. Molecular characterization of eukaryotic algal communities in the tropical phyllosphere based on real-time sequencing of the 18S rDNA gene. BMC Plant Biology, 2018, 18: 1-14.
- Kumar V, Al Momin S, Kumar V V, et al. Distribution and diversity of eukaryotic microalgae in Kuwait waters assessed using 18S rRNA gene sequencing. Plos one, 2021, 16(4): e0250645.
- Xu H, Zhang S, Ma G, et al. 18S rRNA gene sequencing reveals significant influence of anthropogenic effects on microeukaryote diversity and composition along a river-to-estuary gradient ecosystem. Science of the total environment, 2020, 705: 135910.
Featured Resources
How to Identify Microorganisms Quickly and Accurately
MICROBIOME STUDIES: 16S/18S/ITS Amplicon Sequencing or Metagenomics
* For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.



