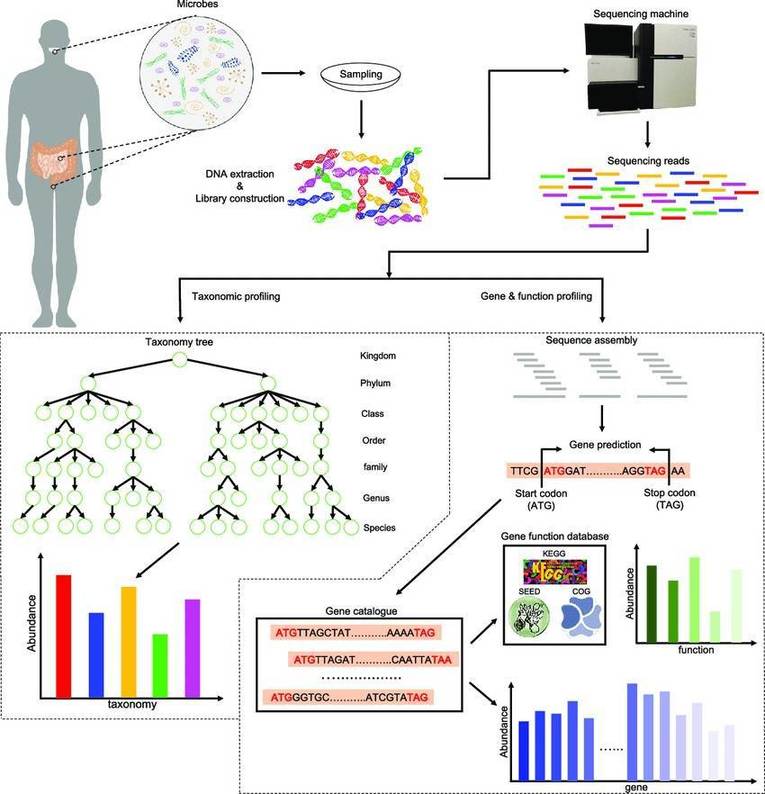
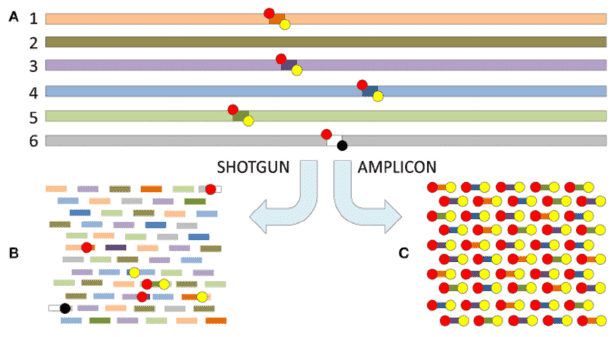
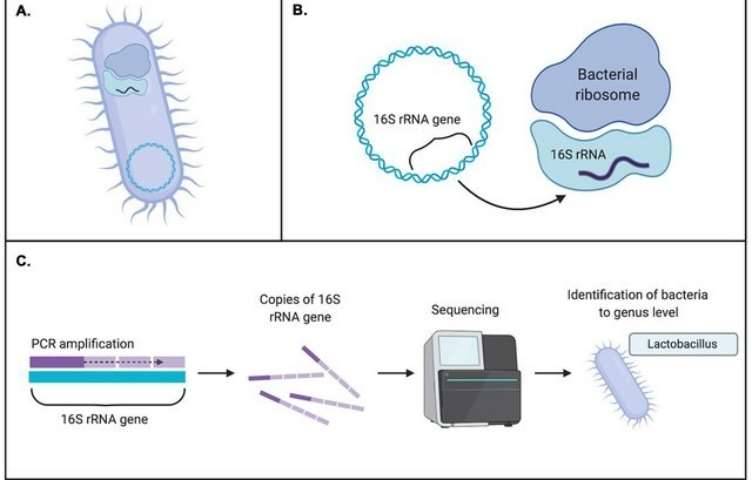
Further to a comprehensive discussion on the principles and applications of metagenomic sequencing (a detailed explanation can be found in our article "Metagenomics Sequencing Overview"), the present exploration intends to elucidate the potent application of metagenomic sequencing across four routine research paradigms. Utilizing specific case illustrations, we aim to demonstrate the strategic leverage of metagenomic sequencing, thereby heightening the rigour of microbial community research.
Scenario One: Insufficient Sample Size for Microbiome Research
In the domain of microbiome research, the diversity of microbial composition across populations is shaped by an array of factors encompassing individual genetics, physiological parameters, health status, lifestyle choices, and ethnic backgrounds. Hence, the appropriateness of sample quantity is fundamentally linked to the dependability of research results. This inevitably raises the question of what should be categorized as an adequate number of samples. Using clinical studies centering on microbial communities as an illustration, it is suggested that a minimum of 100 samples per group are necessary for amplicon sequencing. Nevertheless, procuring a sizeable number of samples for practical research can present as an uphill task due to limitations in project schedules and the pace of sample accumulation, with time constraints further complicating the situation.
In order to uphold the integrity and credibility of published research, the consideration for using metagenomic sequencing methods in microbiome studies often hinges on ensuring adequate sample size. Current standards for such experiments suggest a minimum sample size of at least 50 per experimental group.
Case 1: Inflammation-type dysbiosis of the oral microbiome associates with the duration of COVID-19 symptoms and long COVID
Journal: JCI Insight
Impact Factor: 8.315
Published: October 2021
Research Objective: To examine the relationship between oral microbiota and the duration of long-term COVID-19 symptoms. Study Groups: Early symptom relief (13 cases), persistent symptoms (4 cases), and long-term COVID (10 cases).
Samples: Oropharyngeal swabs collected before treatment.
Research Methodology: Metagenomic sequencing
Research Approach:
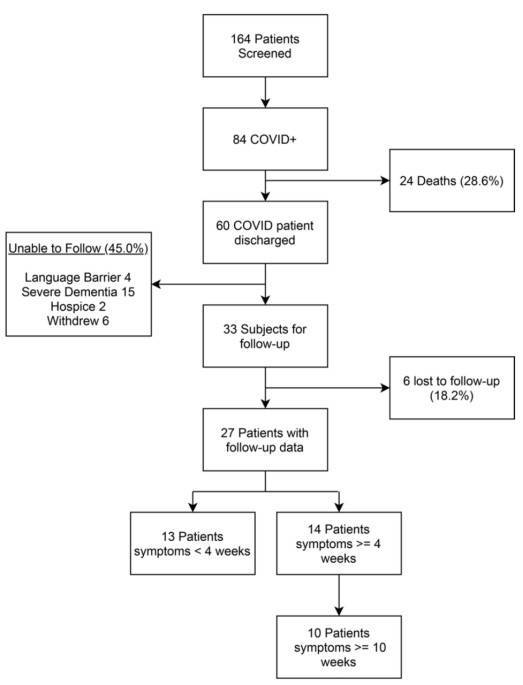
Research Discoveries: Exploring the Link Between Oral Microbiota and Long-Term COVID-19 Symptoms, Deciphering the Possible Role of Functional Imbalances in Oral Microbiota in Persistent COVID-19 Manifestations. Herein, key findings and illustrative representations are advanced for your review.
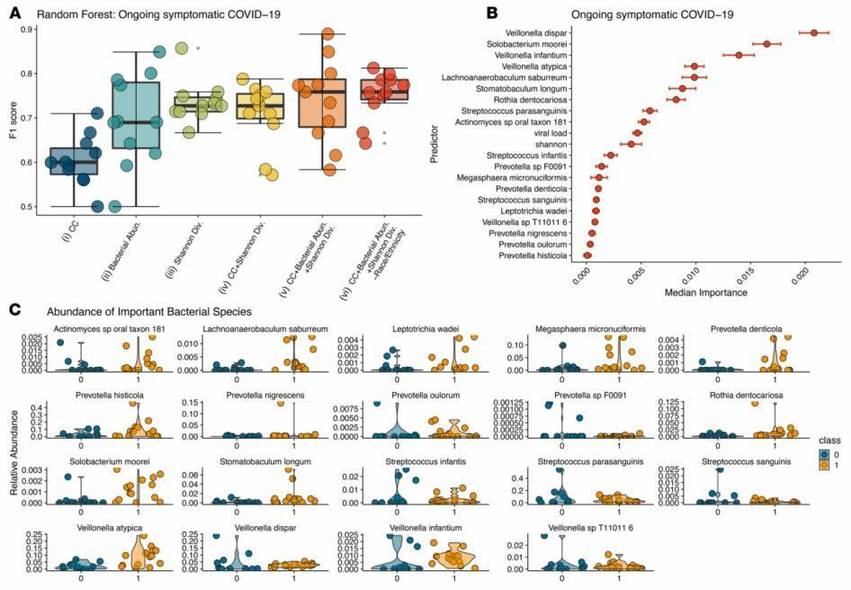 Figure 1: Prediction of Bacterial Abundance in Emerging COVID-19 Symptoms
Figure 1: Prediction of Bacterial Abundance in Emerging COVID-19 Symptoms
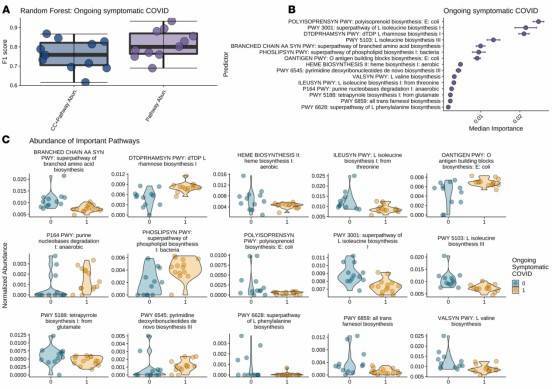 Figure 2: Significantly Associated Metabolic Pathways of Inflammation-Related Bacteria with Persistent COVID-19 Symptoms
Figure 2: Significantly Associated Metabolic Pathways of Inflammation-Related Bacteria with Persistent COVID-19 Symptoms
Case 2: Shotgun Sequencing of the Vaginal Microbiome Reveals Both a Species and Functional Potential Signature of Preterm Birth
Journal: NPJ Biofilms and Microbiomes
Impact Factor: 7.290
Published: November 2020
Research Objective: The research objective seeks to delineate the discrepancies in the composition of vaginal microbiota and gene function between high-risk preterm birth individuals and low-risk control groups. To address this, they employ high-resolution metagenomic sequencing for an extensive investigation.
Study Groups: Women at high risk of preterm birth (35 cases) and low-risk control group (14 cases).
Study Samples: Vaginal swabs
Research Methodology: Metagenomic sequencing
Research Findings: The identification of Lactobacillus iners is closely associated with full-term pregnancy, while other microbial communities are correlated with preterm birth. The functional potential of the microbiota in preterm birth differs from that in full-term pregnancy. Selected results and figures are presented below.
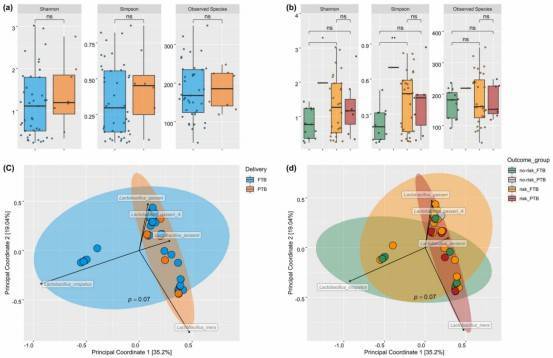 Fig. 1: Species diversity between study groups.
Fig. 1: Species diversity between study groups.
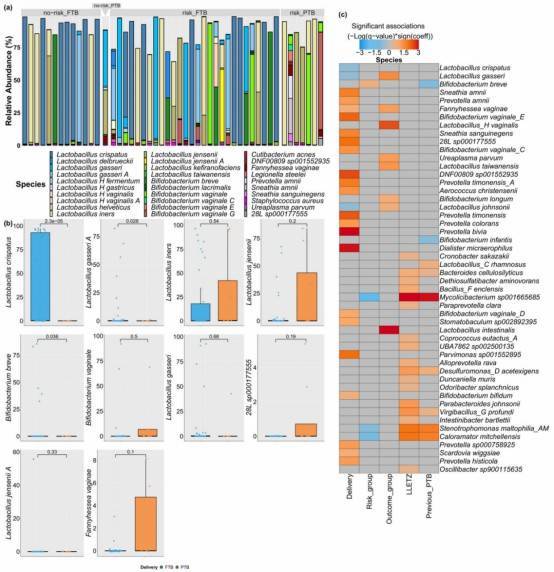 Fig. 2: Species composition across study groups.
Fig. 2: Species composition across study groups.
Scenario Two: What to Do When the Resolution of Microbial Composition Identification is Insufficient?
Through 16S sequencing technology, we can identify microbial communities with accuracy reaching the genus level. However, when attempting to identify at the species level, the resolution of this technique falls short of meeting the demands. Although there are full-length 16S sequencing technologies that have improved accuracy in identifying microbial communities at the species level, limitations persist due to the constraints of existing databases, making it challenging to obtain comprehensive and accurate species information.
In this context, the application of metagenomic sequencing technology becomes particularly crucial. Compared to 16S sequencing technology, metagenomic sequencing offers higher resolution, primarily owing to its reliance on the NR database. Leveraging the rich genetic information within this database, metagenomic sequencing can identify microbial communities at the species level and, in certain instances, even achieve strain-level resolution. Undoubtedly, this capability provides robust support for the exploration of microbial biomarkers with enhanced predictive potential.
Case 3: Compositional and genetic alterations in the gut microbiome of Graves' disease reveal specific diagnostic biomarkers
Journal: ISME J.
Impact Factor: 10.302
Published: November 2021
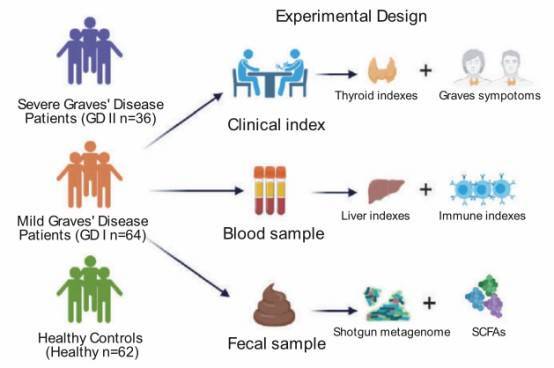
Research Objective: Explore alterations in the classification, genes, pathways, functions, metabolic products, and mutation spectrum of the gut microbiome in Graves' disease (GD) patients. Establish microbial markers for the diagnosis of GD.
Study Groups: Severe Graves' disease patients (GD II, 36 individuals) vs. Mild Graves' disease patients (GD I, 64 individuals) vs. Healthy Control Group (62 individuals)
Sample Type: Stool samples
Research Methods: Metagenomic sequencing + Short-chain fatty acid detection
Research Overview:
Research Findings: Identified a combination of biomarkers, including species, metagenome-assembled genomes (MAGs), genes, and SNPs, whose predictive ability for Graves' disease (GD) surpasses that of any individual biomarker.
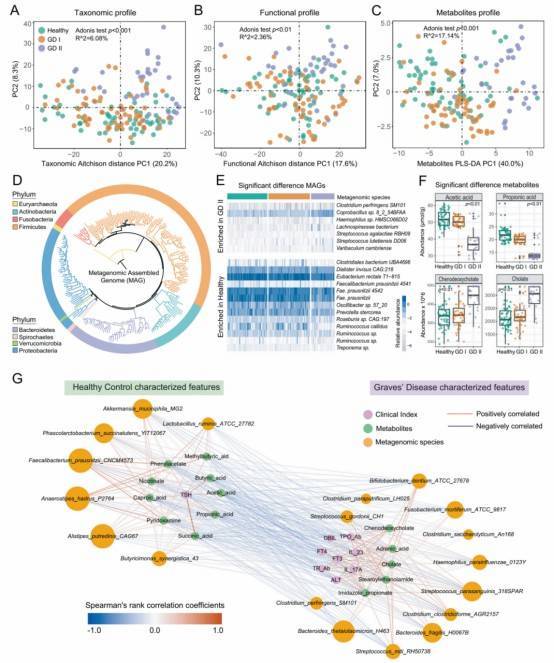 Fig. 1: The Alteration of intestinal microbiome and microbial metabolites in GD patients.
Fig. 1: The Alteration of intestinal microbiome and microbial metabolites in GD patients.
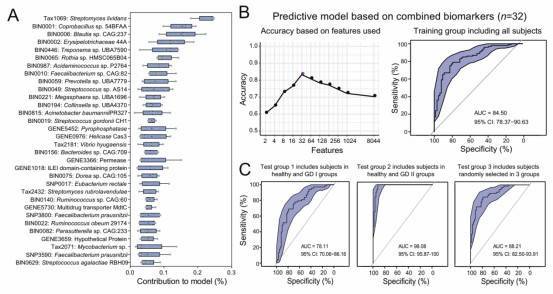 Fig. 2: Identification of GD-associated biomarkers using a machine learning approach and the multi-cohort analysis reveals gut microbiome biomarkers that are specific to GD.
Fig. 2: Identification of GD-associated biomarkers using a machine learning approach and the multi-cohort analysis reveals gut microbiome biomarkers that are specific to GD.
Scenario Three: After Conducting 16S Sequencing, How to Enhance Microbiome Research
In the study of the microbiome, limiting analysis to the diversity of 16S sequencing data is insufficient. To gain a more profound understanding of the relationship between microbial dysbiosis and the onset and progression of diseases, along with their underlying mechanisms, we must delve further into the functional roles played by microbial communities.
To achieve this goal, metagenomic sequencing technology emerges as a potent tool. Through this technique, we can acquire genetic information carried by microbial communities and subsequently analyze the functions and pathways in which these communities are involved. Undoubtedly, this approach significantly elevates the depth and comprehensiveness of our microbiome research.
Case 4: Dysbiosis of human gut microbiome in young onset colorectal cancer
Journal: Nat Commun.
Impact Factor: 14.919
Published: November 2021
Research Objective: Investigating the diagnostic value of the gut microbiota in patients with early-onset colorectal cancer (yCRC).
Research Groups: yCRC vs. Late-onset (oCRC) vs. Control, details provided in the flowchart.
Sample Type: Stool samples
Research Methods: 16S sequencing (1038 cases) + Metagenomic sequencing (200 cases)
Research Overview:
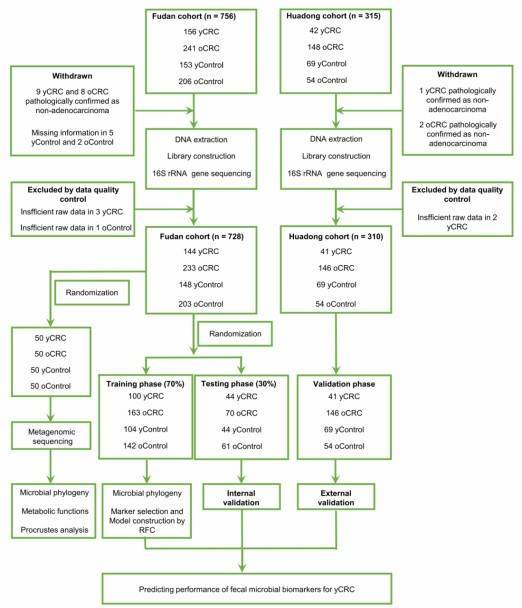
Research Findings: Through metagenomic sequencing, distinct bacterial genera were found to be significantly enriched in both young-onset colorectal cancer (yCRC) and older-onset colorectal cancer (oCRC) patients. In yCRC, unique bacterial metabolic functional characteristics were discovered. Furthermore, a diagnostic model was constructed based on these microbial markers.
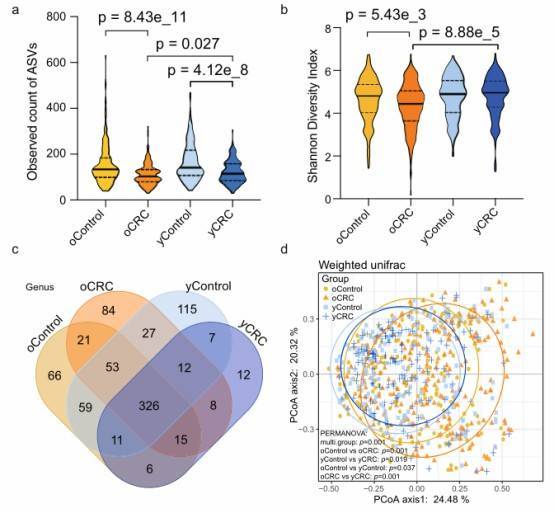 Fig. 1: Bacterial diversity of the fecal microbiota associated with yCRC and oCRC.
Fig. 1: Bacterial diversity of the fecal microbiota associated with yCRC and oCRC.
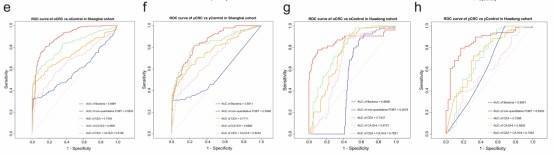 Fig. 2: Independent validation and diagnostic performance of fecal microbial markers for yCRC and oCRC.
Fig. 2: Independent validation and diagnostic performance of fecal microbial markers for yCRC and oCRC.
Scenario Four: Enhancing Microbiome Research Beyond Metagenomics Sequencing
The scope of microbiome research has vital importance, not only revealing the intricacies of microbial communities but also enhancing our knowledge regardiing the interplay between these communities and their host organisms. The influence of microbial communities on their hosts primarily manifests through the metabolites they generate. Hence, for a comprehensive understanding of microbial-host interactions, it is necessary to consider both the composition and dynamic variations of microbial communities, along with examining the consequences of alterations in microbial metabolite production on the host organism.
Additionally, attention should be directed towards variations in host blood metabolomics, which may correlate with changes in microbial metabolomics. Depending on the specific research objectives and subjects, a comprehensive investigation might involve examining changes in the host's transcriptome and proteome. This holistic approach allows for a more profound understanding of the mechanisms governing the interactions between microbial communities and their hosts.
Case Five: Disruptions in the Gut Microbiome and Metabolome in Children with Calcium Oxalate Kidney Stone Disease
Journal: American Society of Nephrology
Impact Factor: 10.121
Published: June 2020
Research Objective: To investigate the relationship between the composition and functionality of the gut microbiome and early-onset calcium oxalate kidney stone disease.
Study Groups: Kidney stone patients (44 cases) and a control group (44 cases).
Study Samples: Stool samples.
Research Methods: Metagenomic sequencing coupled with untargeted metabolomics.
Research Findings: The absence of gut microbiota is associated with disruptions in the metabolome, suggesting that the metabolome may serve as an upstream determinant in the pathogenesis of early-onset calcium oxalate kidney stone disease.
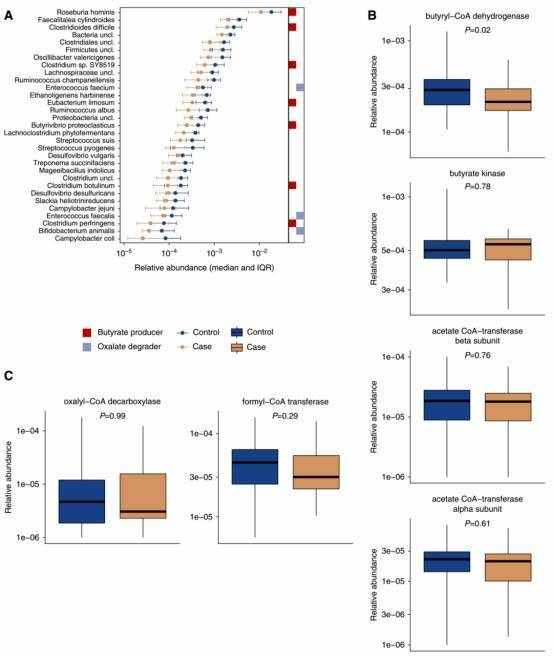 Figure 1. The relative abundance of 32 bacterial taxa and the abundance of the bacterial gene butyryl-CoA dehydrogenase were lower among participants with kidney stone disease.
Figure 1. The relative abundance of 32 bacterial taxa and the abundance of the bacterial gene butyryl-CoA dehydrogenase were lower among participants with kidney stone disease.
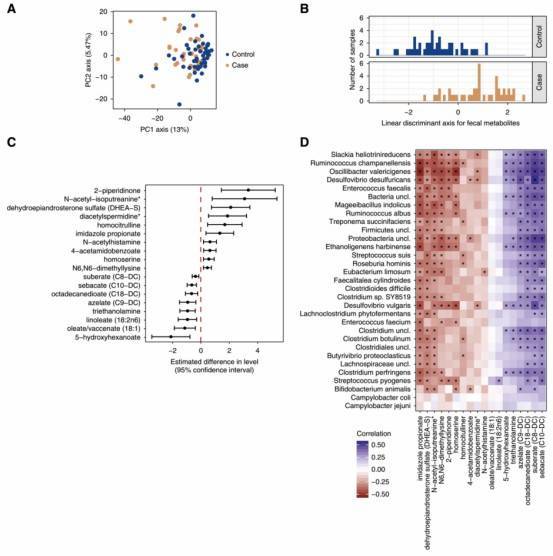 Figure 2. The fecal metabolome is distinct in participants with kidney stone disease.
Figure 2. The fecal metabolome is distinct in participants with kidney stone disease.
Case Study 6: Alterations in Fecal Microbiomes and Serum Metabolomes of Fatigued Patients With Quiescent Inflammatory Bowel Diseases
Journal: Clin Gastroenterol Hepatol.
Impact Factor: 11.382
Published: March 2021
Research Objective: Investigate changes in the fecal microbiome, serum metabolome, and proteome of fatigued/non-fatigued Crohn's disease patients to elucidate the mechanisms underlying fatigue symptoms in Crohn's disease.
Study Groups: 106 cases of quiescent Crohn's disease, 60 cases of ulcerative colitis
Samples: Fecal samples, serum
Methods: Metagenomic sequencing + Untargeted metabolomics + Olink proteomics
Research Findings: An analysis of the fecal microbiota, serum metabolome, and proteome of 166 IBD patients revealed associations between changes in serum metabolites, fecal microbiota, and fatigue.
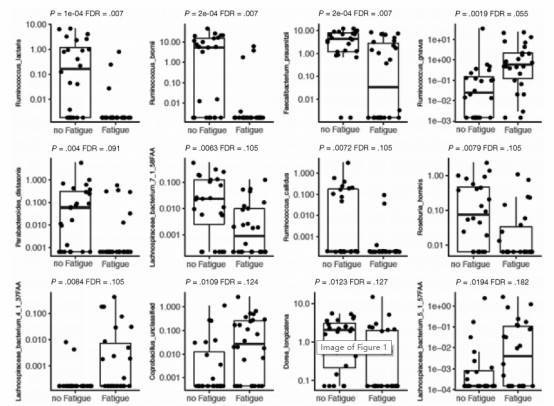 Figure 1. Species-level differences in the microbiome between patients with quiescent inflammatory bowel disease
Figure 1. Species-level differences in the microbiome between patients with quiescent inflammatory bowel disease
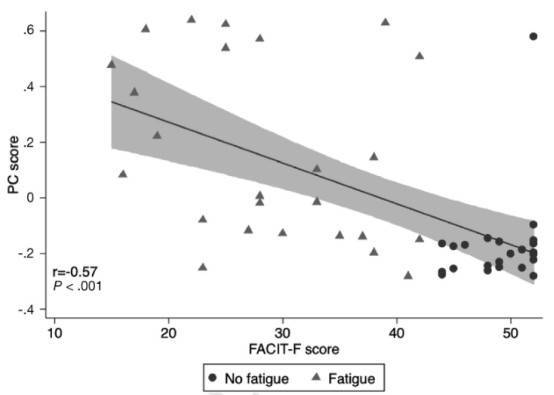 Figure 2. Correlation of the fatigue microbiome score on principal component (PC) analysis with quantitative burden of fatigue.
Figure 2. Correlation of the fatigue microbiome score on principal component (PC) analysis with quantitative burden of fatigue.
Case Study 7: Alterations of the Gut Ecological and Functional Microenvironment in Different Stages of Multiple Sclerosis
Journal: PNAS
Impact Factor : 11.205
Published: September 2020.
Objective: The study aims to elucidate the microbial composition and functional differences in the gut microbiome of individuals at different stages of Multiple Sclerosis (MS).
Research Groups: The research encompasses 62 cases of RRMS (Relapsing-Remitting MS), 15 cases of SPMS (Secondary Progressive MS), 21 cases of Atypical MS, 20 cases of NMOSD (Neuromyelitis Optica Spectrum Disorder), and includes a control group of 55 healthy individuals.
Sample Collection: Fecal samples were collected as the primary source for the investigation.
Methodology: The study employed a comprehensive approach, utilizing 16S sequencing, metagenomic sequencing, and targeted metabolomics focused on Short-Chain Fatty Acids (SCFA) and sulfur metabolism.
Research Findings:
In the course of distinct Multiple Sclerosis (MS) progression trajectories, significant alterations were observed in the intestinal ecological and functional microenvironment. Particularly noteworthy were the reduced biosynthesis of short-chain fatty acids in Relapsing-Remitting MS (RRMS) and elevated oxidative levels in Secondary Progressive MS (SPMS). These findings underscore the intricate dynamics of the gut milieu during different stages of MS development.
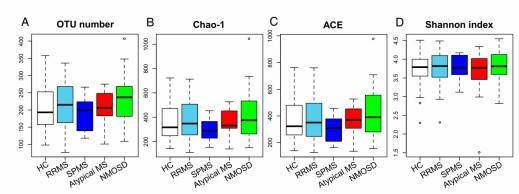 Fig. 1. Gut microbiome alpha-diversities in the five subject groups.
Fig. 1. Gut microbiome alpha-diversities in the five subject groups.
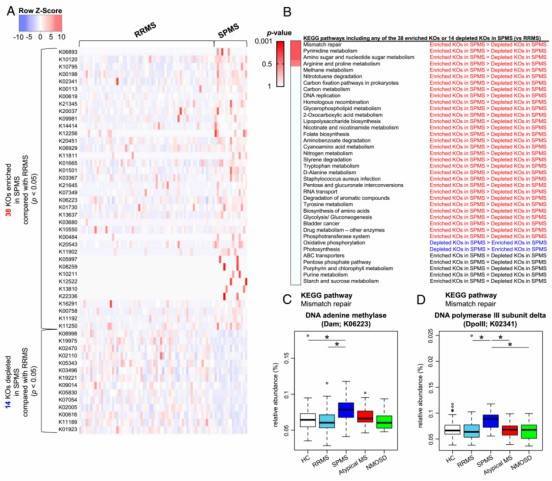 Fig. 2. Differences in functional profile of gut microbiome between RRMS and SPMS.
Fig. 2. Differences in functional profile of gut microbiome between RRMS and SPMS.
References
- Takewaki D, et al. Alterations of the gut ecological and functional microenvironment in different stages of multiple sclerosis. Proc Natl Acad Sci U S A. 2020 Sep 8;117(36):22402-22412.
- Chu W, et al. Metagenomic analysis identified microbiome alterations and pathological association between intestinal microbiota and polycystic ovary syndrome. Fertil Steril. 2020 Jun;113(6):1286-1298.e4.
- Borren NZ, et al. Alterations in Fecal Microbiomes and Serum Metabolomes of Fatigued Patients With Quiescent Inflammatory Bowel Diseases. Clin Gastroenterol Hepatol. 2021 Mar;19(3):519-527.e5.
- Denburg MR, et al. Perturbations of the Gut Microbiome and Metabolome in Children with Calcium Oxalate Kidney Stone Disease. J Am Soc Nephrol. 2020 Jun;31(6):1358-1369.
- Yang Y, et al. Dysbiosis of human gut microbiome in young-onset colorectal cancer. Nat Commun. 2021 Nov 19;12(1):6757.
- Haran JP, et al. Inflammation-type dysbiosis of the oral microbiome associates with the duration of COVID-19 symptoms and long COVID. JCI Insight. 2021 Oct 22;6(20):e152346.
- Feehily C, et al. Shotgun sequencing of the vaginal microbiome reveals both a species and functional potential signature of preterm birth. NPJ Biofilms Microbiomes. 2020 Nov 12;6(1):50.
- Zhu Q, et al. Compositional and genetic alterations in Graves' disease gut microbiome reveal specific diagnostic biomarkers. ISME J. 2021 Nov;15(11):3399-3411.