CD Genomics employs next-generation high-throughput sequencing technology to sequence the transcriptomes of prokaryotes (bacteria and archaea), enabling comprehensive and rapid acquisition of the complete transcriptomic information (including mRNA and non-coding RNA) from individual microbial colonies or microbial communities.
Our Advantages:
- Comprehensive Coverage: Our platform captures both coding and non-coding regions, providing a complete transcriptome view including sRNAs, antisense transcripts, and novel regulatory elements.
- High Sensitivity and Accuracy: Advanced rRNA depletion and high-throughput sequencing accurately detect even low-abundance transcripts, essential for understanding bacterial responses and regulatory genes.
- Versatility Across Strains: Suitable for various bacterial strains, our service includes de novo transcriptome assembly for strains without reference genomes, facilitating the study of uncharted species.
- Robust Bioinformatics Support: We offer extensive bioinformatics services, from initial data processing to advanced analyses like differential gene expression and pathway enrichment, ensuring high-quality, publication-ready data.
What is RNA-seq Analysis of Bacteria
RNA-seq analysis is an advanced technique for studying the bacterial transcriptome in detail. Unlike eukaryotic mRNA, bacterial mRNA lacks a poly(A) tail, requiring specialized methods for library preparation. This technique enables precise measurement of gene expression, discovery of new transcripts, and investigation of gene regulation. In prokaryotes, where transcription and translation happen simultaneously, RNA-seq captures gene expression in specific environments, helping identify key functional genes in metabolic and regulatory pathways.
The bacterial transcriptome is complex, featuring polycistronic mRNAs that encode multiple proteins. This complexity, along with the short lifespan of bacterial mRNAs and potential overlaps, demands specialized analysis tools. RNA-seq provides insights into operon structures, the role of non-coding RNAs, and the dynamics of gene expression, which are crucial for understanding bacterial physiology and pathogenicity.
Introduction to Prokaryotic RNA Sequencing
Prokaryotic RNA sequencing is a powerful, high-throughput technology designed to capture and analyze the RNA transcripts present in bacterial cells at a specific time point. Unlike eukaryotic RNA sequencing, which frequently relies on the presence of a poly(A) tail for mRNA isolation, prokaryotic RNA sequencing employs rRNA depletion methodologies to enrich for mRNA and other non-coding RNAs. This strategy enables a comprehensive analysis of the bacterial transcriptome, encompassing coding and non-coding regions, antisense transcripts, and regulatory elements.
At CD Genomics, our prokaryotic RNA sequencing platform leverages cutting-edge Illumina HiSeq (PE150) and PacBio SMRT systems to deliver high-resolution insights into bacterial gene expression. This platform is versatile, catering to both well-established strains with reference genomes and novel strains lacking genomic information. The data generated through this technology can be applied to a variety of research pursuits, including gene function annotation, differential gene expression analysis, and the elucidation of key regulatory networks within bacterial systems.
Applications of Prokaryotic RNA Sequencing
Prokaryotic RNA sequencing has a wide range of applications across various fields:
- Microbial Physiology and Metabolism: RNA-seq enables the exploration of metabolic pathways and regulatory networks in bacteria, aiding studies of industrially significant species.
- Pathogen-Host Interactions: Analyzing pathogenic bacteria's transcriptomes reveals infection mechanisms and antibiotic resistance, informing therapeutic and vaccine development.
- Environmental Microbiology: Prokaryotic RNA sequencing monitors microbial communities in various habitats, enhancing understanding of bacterial adaptation and ecosystem interactions.
- Biotechnology and Synthetic Biology: RNA-seq aids in engineering bacteria for desired traits by elucidating gene expression patterns, optimizing metabolic pathways, and enhancing bioproduct production.
Service Specifications
Prokaryotic RNA Sequencing Workflow
The prokaryotic RNA sequencing workflow at CD Genomics involves several essential steps. It begins with total RNA extraction and quality assessment, followed by rRNA depletion to enrich for mRNA and non-coding RNAs. The remaining RNA is converted to cDNA with strand specificity using dUTP, and then amplified with sequencing adapters. Sequencing is performed using either Illumina for short reads or PacBio for longer reads. Finally, CD Genomics conducts bioinformatics analysis for quality control, transcript assembly, differential gene expression analysis, and functional annotation, utilizing advanced tools like Rockhopper 2 to address the complexities of prokaryotic transcriptomes.

Technical Parameters
- HiSeq4000, PE150, >4G clean data
- PacBio SMRT systems
- Nanopore platform
Note: The above content includes only a portion of the bioinformatics analysis. For more information or to customize the analysis, please contact us directly.
Bioinformatics Analysis
Our bioinformatics analysis includes these parts: read QC and assembly, expression analysis, structure analysis, and advanced analysis. For more detailed bioinformatics analysis, please refer to the following table.
| Read QC & Assembly |
| Quality assessment of raw data |
Contamination detection |
| Mapping to the reference genome |
De novo assembly |
| Structure analysis |
| Prediction of novel transcripts |
UTR analysis and annotation |
| SNP and indel analysis |
Operon analysis |
| sRNA analysis |
Prediction of antisense transcripts |
| Gene fusion discovery |
|
| Expression analysis |
| GO/KEGG enrichment analysis |
Cluster analysis |
| Gene expression quantification |
New gene sequence annotation |
| PCA |
Alternative splicing analysis |
| Advanced analysis |
| Metabolic pathway integration analysis |
Gene co-expression network analysis |
| Protein interaction network analysis |
mRNA-sRNA co-expression network analysis |
| Correlation analysis |
|
Reference-based prokaryotic transcriptome analysis workflow:
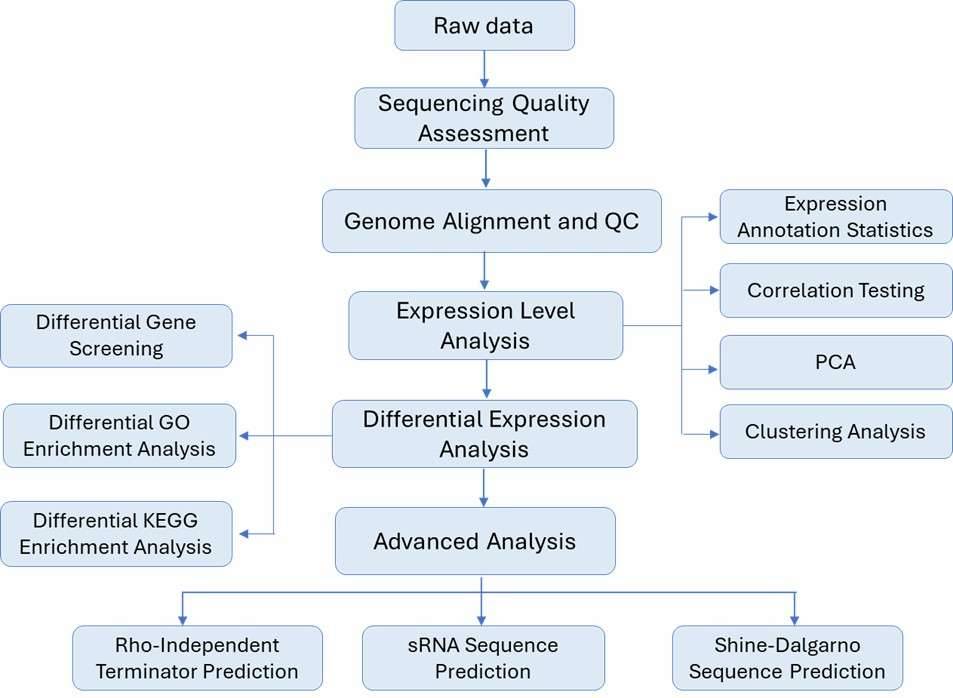
Sample Requirement
| Sample Type |
Recommended Quantity |
| Total RNA |
≥ 1 µg |
| Cells |
≥ 1×107 |
- OD A260/A280 ratio ≥ 1.8, A260/230 ratio≥ 1.8, RIN ≥ 6
- All total RNA samples should be DNA-free
Note: If you wish to obtain more accurate and detailed information regarding sample requirements, please feel free to contact us directly.
Deliverables
- Raw sequencing data (FASTQ)
- Clean data
- Trimmed and stitched sequences (fasta)
- Quality-control dashboard
- Sample contamination report
- Statistic data
- Your designated bioinformatics result report
Please feel free to reach out if you have any further inquiries or require additional information.
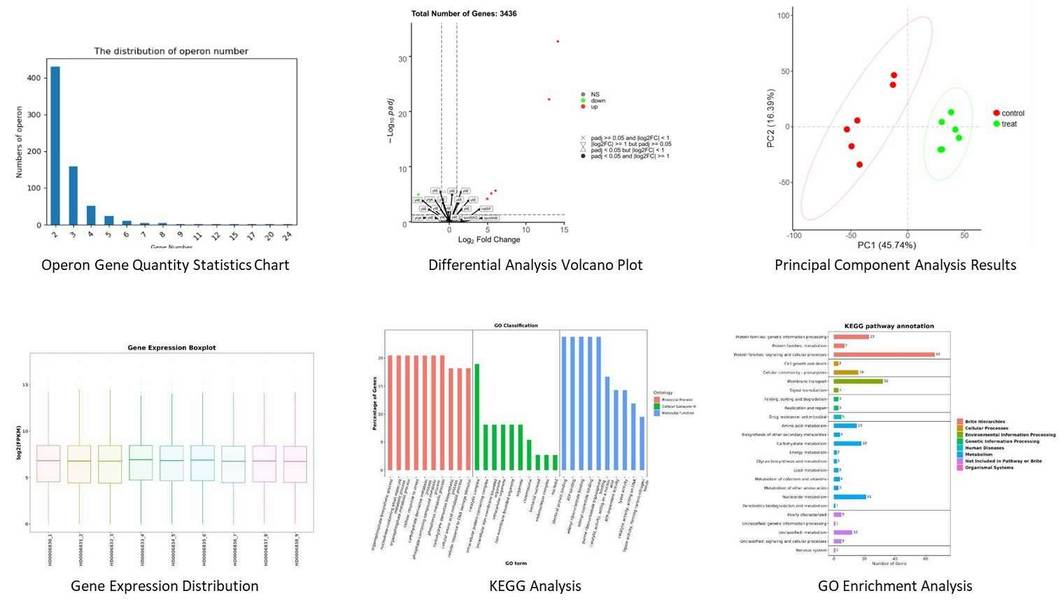

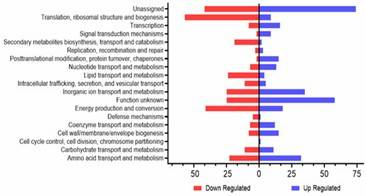 Figure 1. A. baumannii WT and YhaK Tn Transcriptome analysis: KEGG gene ontology groups.
Figure 1. A. baumannii WT and YhaK Tn Transcriptome analysis: KEGG gene ontology groups.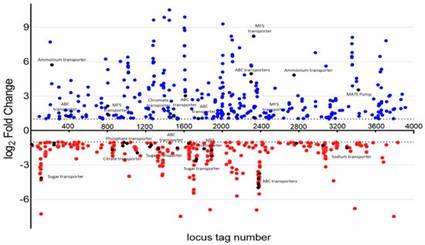 Figure 2. A. baumannii WT and YhaK Tn Transcriptome analysis: differentially expressed genes.
Figure 2. A. baumannii WT and YhaK Tn Transcriptome analysis: differentially expressed genes.