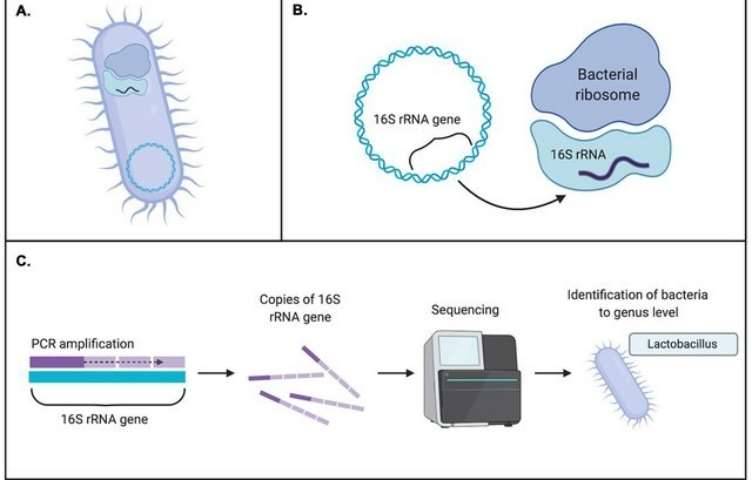
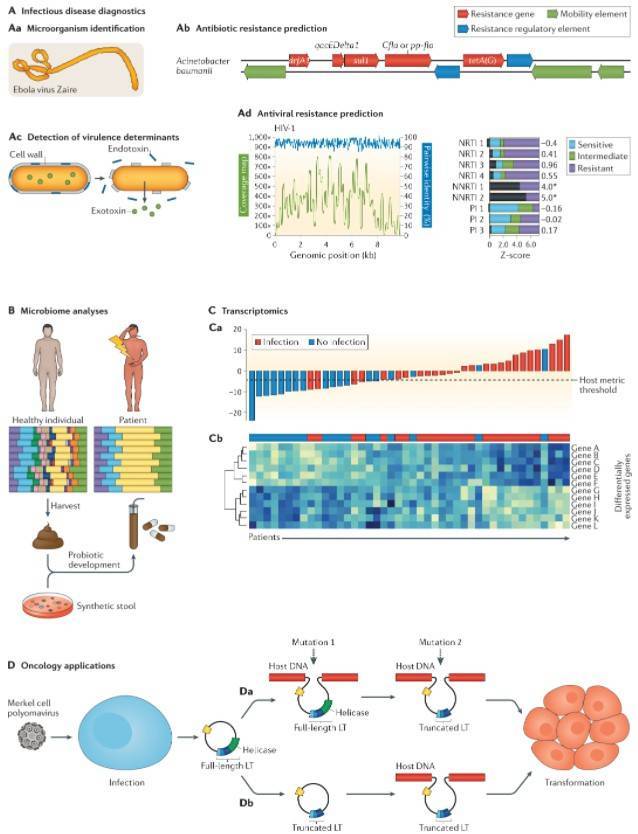
In the realm of microbial community investigation, particularly concerning the analysis of gut microbiota, a common initial approach involves the application of amplicon sequencing methods, such as 16S rRNA gene sequencing. The utilization of 16S rRNA gene sequencing stands out due to its cost-effectiveness and the accessibility of easily interpretable data, rendering it an optimal starting point for microbial community research.
Nevertheless, an alternative tool in microbial community research, metagenomic sequencing, is frequently overlooked. Part of the reason may stem from its relatively higher cost. Simultaneously, the sufficiency of 16S rRNA gene sequencing for certain research demands has led to an underestimation of the significance of metagenomic sequencing.
In the realm of microbial community exploration, the employment of high-resolution metagenomic sequencing techniques assumes a pivotal role. This methodology not only elevates species resolution to the level of individual strains but also delves into the genetic functional information within microbial communities. This groundbreaking technology aids us in a more comprehensive and precise delineation of microbial species composition. Moreover, leveraging the genetic information harbored by these communities enables a more profound understanding of their functional characteristics.
What is Metagenomics Sequencing?
Metagenome, also referred to as Environmental or Community Genome, encompasses the entirety of genetic material from all microorganisms within a specific habitat, encompassing both cultivable and non-cultivable microbial genomes. Metagenomics, employing high-throughput sequencing platforms such as NGS or PacBio, focuses on the genomic analysis of microbial populations in a given environment. Beyond elucidating microbial diversity, population structures, and evolutionary relationships, metagenomics allows for a more in-depth exploration of functional activities within microbial communities, their cooperative interactions, and their relationships with the environment, uncovering potential biological significance. Currently, the primary areas of research in metagenomics include the investigation of viral metagenomics and bacterial metagenomics.
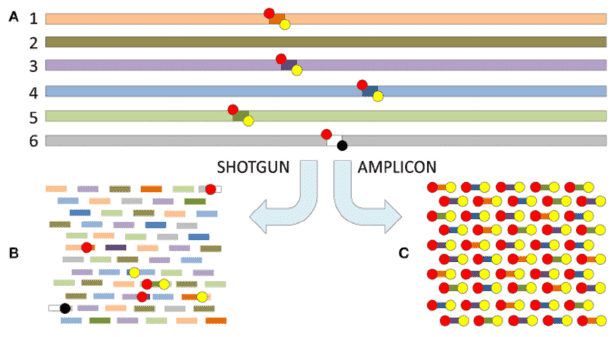
The basic outline of metagenomic sequencing and data processing. (Xuegong Zhang et al,. 2016)
Advantages of Metagenomic Sequencing
In contrast to traditional microbial research methods, metagenomic sequencing technology successfully circumvents the challenges posed by numerous uncultivable microorganisms and the detection limitations associated with trace amounts of bacteria. As a result, it has found widespread application in the field of environmental microbiology. Metagenomic sequencing not only accurately identifies the species composition of bacterial communities in a given sample but also enables a more in-depth analysis of the composition of other microbial communities. This is achieved by sequencing the entire genome of microbial communities using metagenomic sequencing, coupled with the utilization of information-rich gene databases such as the NR database for species identification. Consequently, metagenomic sequencing exhibits a higher species resolution, capable of reaching the level of individual strains or even sub-strains.
Furthermore, the genetic information on microbial communities obtained through metagenomic sequencing allows us to delve into the functionality of these genes and the metabolic pathways in which they participate. This, in turn, facilitates a deeper understanding of the interactions between microbial communities and their hosts. These distinctive advantages render metagenomic sequencing instrumental in exploring research at the functional and mechanistic levels, a feat unattainable through amplicon sequencing alone.
In contrast to NGS methods, Long Read metagenomics, utilizing the PacBio SMRT/Nanopore platform's long-read technology, offers a more precise coverage of gene intervals or specific regions. This not only reduces assembly errors but also authentically reflects the intricate composition of microbial communities. Such technological advancements enhance the resolution of microbial community identification, laying a robust foundation for subsequent functional studies.
By correcting the original error rate of PacBio sequencing/Nanopore sequencing, we attain high-quality Circular Consensus Sequencing (CCS) reads with a minimum accuracy exceeding 99%. This enables comprehensive coverage of genes or specific regions for the majority of microorganisms, allowing for microbial community composition analysis, functional gene annotation, and metabolic pathway exploration without the need for assembly.
Building upon high-quality CCS reads, we employ a Long Read assembly strategy. This approach enhances specificity by incorporating reads from diverse microorganisms, effectively preventing cross-assembly between different individuals. This precision allows us to accurately reconstruct the composition and functional gene information of microbes in the environment, facilitating in-depth exploration of microbial community data.
Long-read sequencing as represented by single-molecule real-time (SMRT) sequencing (Gavin Douglas 2019)
In the course of research endeavors, if one aspires to delve deeply into the intricacies of the microbial community, particularly the interplay mechanisms between the gut microbiota and the host, metagenomic sequencing undoubtedly emerges as the method of choice. Simultaneously, when precise identification of core microbial species closely associated with diseases or specific phenotypes is requisite, metagenomic sequencing equally stands out as the most appropriate technological approach.
In practical application, metagenomic sequencing and amplicon sequencing often complement each other. Amplicon sequencing is primarily suitable for analyzing microbial community diversity in large-scale samples, while metagenomic sequencing focuses on in-depth exploration of microbial gene functionality and pathways within key samples. This combination not only contributes to a more comprehensive species identification but also provides deeper insights into microbial functional information. For specific scenarios where metagenomic sequencing finds application, a more detailed understanding can be obtained by referring to the article "How to Use Metagenomic Sequencing."
In human pathogenic microorganisms, over 70% of viruses originate from animal transmission, while more than 99% of microorganisms in nature remain unidentified. Rapid detection in the field of microbiology is crucial, not only relying on clinical experiential judgments. Traditional microbial detection methods, including smears, cultures, serological examinations, PCR, among others, suffer from drawbacks such as prolonged processing times and limited detection scopes. In contrast, metagenomic high-throughput sequencing (NGS) technology offers advantages such as shorter processing times and broader detection ranges.
Metagenomics Next-Generation Sequencing (mNGS): This technique utilizes second-generation sequencing platforms for rapid sequencing of nucleic acid sequences in samples. Subsequently, alignment with the genome sequences of various species allows for the determination of the types and proportions of microorganisms present in the sample.
Metagenomics holds an indispensable position in elucidating the intricacies of the human gut microbiome. It contributes significantly to the comprehensive cataloging of microbial diversity, discovery of novel genes, and identification of functional microbial imbalances that might lie at the root of human intestinal disorders. Metagenomics provides us with an enriched understanding of the range and functions of the microbiota present in the human gut. Indeed, it sheds light on essential physiological processes like digestion, immune response, and metabolism that these microbes mediate.
By harnessing the insights garnered through metagenomic analysis, researchers are empowered to examine the complex interplay between the human body and its microbial inhabitants. This extends beyond simply charting the relationship between the human body and gut microbiota; it provides a valuable gateway to unveil the reverberating effects of these relationships on broader health outcomes in humans.
Furthermore, employing metagenomics to study transformations in the gut microbiome paves the way for an enhanced understanding of the correlation between microbiota and overall health, thus aiding in the prognosis of ailments linked to the gastrointestinal tract. Leveraging multiple technologies in unison such as 16S rRNA sequencing, transcriptome scrutiny, and whole-genome sequencing gives way to the identification of key associations between fluctuations in the gut microbiota landscape and specific gastrointestinal disorders such as inflammatory bowel disease and colorectal cancer. This synergistic approach offers an indispensable source of comprehensive and meticulous information, which can be tailored into specific clinical diagnoses and treatment methodologies.
Metagenomics in Oral Disease Research
Oral diseases, such as dental caries and periodontal pathologies, are common health conditions within the human population. Numerous microbial species co-inhabit a multifaceted microbial biofilm, commonly referred to as dental plaque. Employing metagenomic analysis to extract DNA information from the oral microbiota offers an enriched understanding of the oral microbial community's composition. It provides crucial insights into the genes and functions of microbes, clarifies the contributions of diverse microbial species to oral health and pathologies, and investigates their intricate interactions. Notably, metagenomics provides the potential to recognize not only known pathogens but also discover novel ones. Such an approach serves as an invaluable tool in expanding our understanding of mechanisms underpinning oral diseases, including dental caries and oral malignancies.
The utilization of metagenomic approaches to monitor antimicrobial resistance (AMR) in wastewater stands as an indispensable choice for investigating the dissemination of environmental resistance. By surveilling the distribution and abundance of AMR in urban wastewater globally, a comprehensive understanding of the levels and types of AMR in specific populations or geographical regions can be obtained. This aids in identifying resistance hotspots, tracking the spread of resistant strains, and assessing the effectiveness of interventions and policies aimed at controlling AMR. Through metagenomic analysis of AMR in urban wastewater, valuable insights are provided to public health authorities and policymakers for formulating evidence-based strategies concerning antimicrobial drug management, infection control, and mitigation of antimicrobial resistance.
Metagenomics, by unveiling microbes that elude cultivation in laboratory settings, has significantly propelled the advancement of microbiology and biotechnology. The versatility of metagenomics across various fields is immense, with the applications highlighted in this article representing only a fraction of its potential. With the continuous development and progress of technology, it is anticipated that the cost of metagenomics will decrease, the procedures and analyses will become more refined, and its applications will broaden. For further insights into the myriad applications of metagenomics, please refer to the review article: "Clinical Applications of High-Throughput Sequencing Technologies in Metagenomics: Current Status, Challenges, and Prospects."
References
Wang, W.L., et al., Application of metagenomics in the human gut microbiome. World J Gastroenterol, 2015. 21(3): p. 803-14.2.
Ko, K.K.K., K.R. Chng, and N. Nagarajan, Metagenomics-enabled microbial surveillance. Nat Microbiol, 2022. 7(4): p. 486-496.
Xu, P. and J. Gunsolley, Application of metagenomics in understanding oral health and disease. Virulence, 2014. 5(3): p. 424-32.4.
Hovi, T., et al., Role of environmental poliovirus surveillance in global polio eradication and beyond. Epidemiol Infect, 2012. 140(1): p. 1-13.5.
Tharak, A., et al., Longitudinal and Long-Term Wastewater Surveillance for COVID-19: Infection Dynamics and Zoning of Urban Community. Int J Environ Res Public Health, 2022. 19(5).6.
Hendriksen, R.S., et al., Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat Commun, 2019. 10(1): p. 1124.
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to for inquiries.
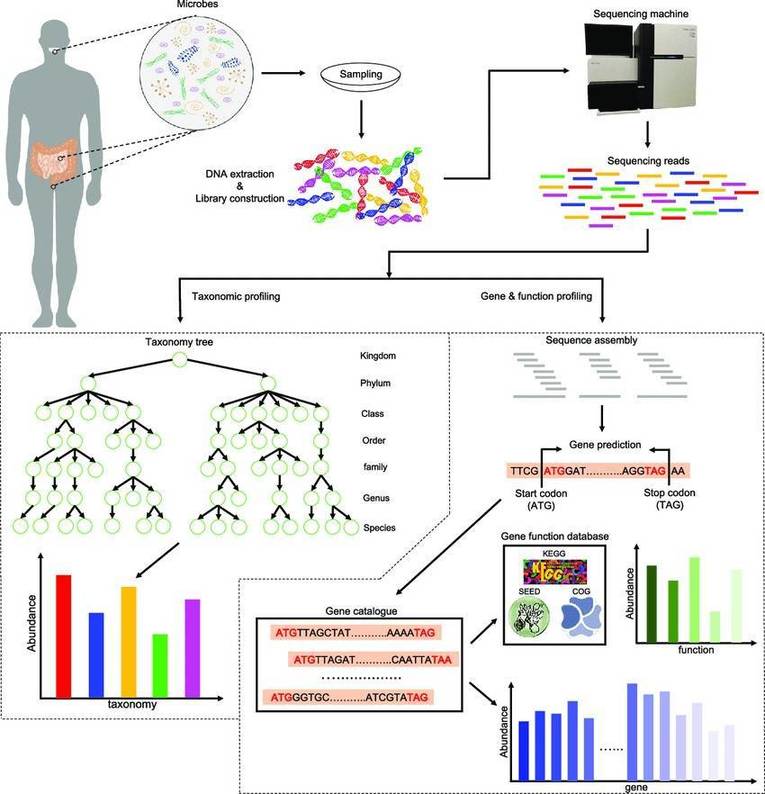 The basic outline of metagenomic sequencing and data processing. (Xuegong Zhang et al,. 2016)
The basic outline of metagenomic sequencing and data processing. (Xuegong Zhang et al,. 2016)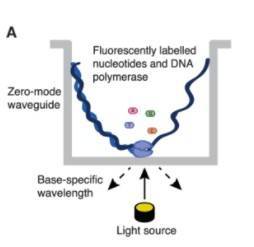 Long-read sequencing as represented by single-molecule real-time (SMRT) sequencing (Gavin Douglas 2019)
Long-read sequencing as represented by single-molecule real-time (SMRT) sequencing (Gavin Douglas 2019)