




We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
In the ongoing interaction between humans and their environment, epigenetics is emerging as a pivotal factor in deciphering the adaptive mechanisms of life. This dynamic interplay encompasses a range of influences, from the "remote manipulation" of host genes by metabolites derived from intestinal flora to the transgenerational DNA methylation memory triggered by pesticide exposure. These environmental signals exert a profound influence on the occurrence of diseases and the evolution of species, with their effects being mediated through epigenetic reprogramming. However, two major challenges exist in analyzing this process: the spatial and temporal heterogeneity of environmental exposures, and the multi-omics integration of microbiome-host interactions. Relying on whole genome methylation sequencing (WGBS), single-cell ATAC-seq, and multi-omics association analysis, we are able to accurately capture the epigenetic fingerprints under environmental stresses and provide novel strategies for agricultural stress breeding and disease prevention.
The dual role of environmental stresses and microbiome metabolites is redefining the boundaries of epigenetics. Flora metabolites, such as short-chain fatty acids (SCFAs), directly modify host DNA methylation, while dietary factors, such as folate deficiency, trigger global epigenetic dysregulation by interfering with one-carbon metabolism. Utilizing WGBS with Human DNA Methylation Microarray, the investigators ascertained that diminished levels of methylation in the promoter region of the IL-10 gene within the colonic mucosa of individuals adhering to a high-fiber diet exhibited a significant and negative correlation with butyric acid concentration. The quantification of this environmental-epigenetic association furnishes a molecular target for precision nutritional interventions.
Gut Microbiota Metabolites: SCFAs as Epigenetic Mediators
Butyric acid, a byproduct of intestinal flora, has been shown to regulate the expression of anti-inflammatory genes, such as Foxp3, by inhibiting histone deacetylases (HDACs) and increasing histone H3K9 acetylation levels. To parse this process, the team used MeDIP-Seq in conjunction with macro-genome sequencing and found that the abundance of anaphylatoxin gates was negatively correlated with the degree of methylation of the TNF-α gene in host peripheral blood mononuclear cells. Integration of RNA-seq and epigenomic data analysis further confirmed that butyric acid could specifically reduce the level of 5mC modification of inflammation-related genes by up-regulating DNA demethylase TET2.
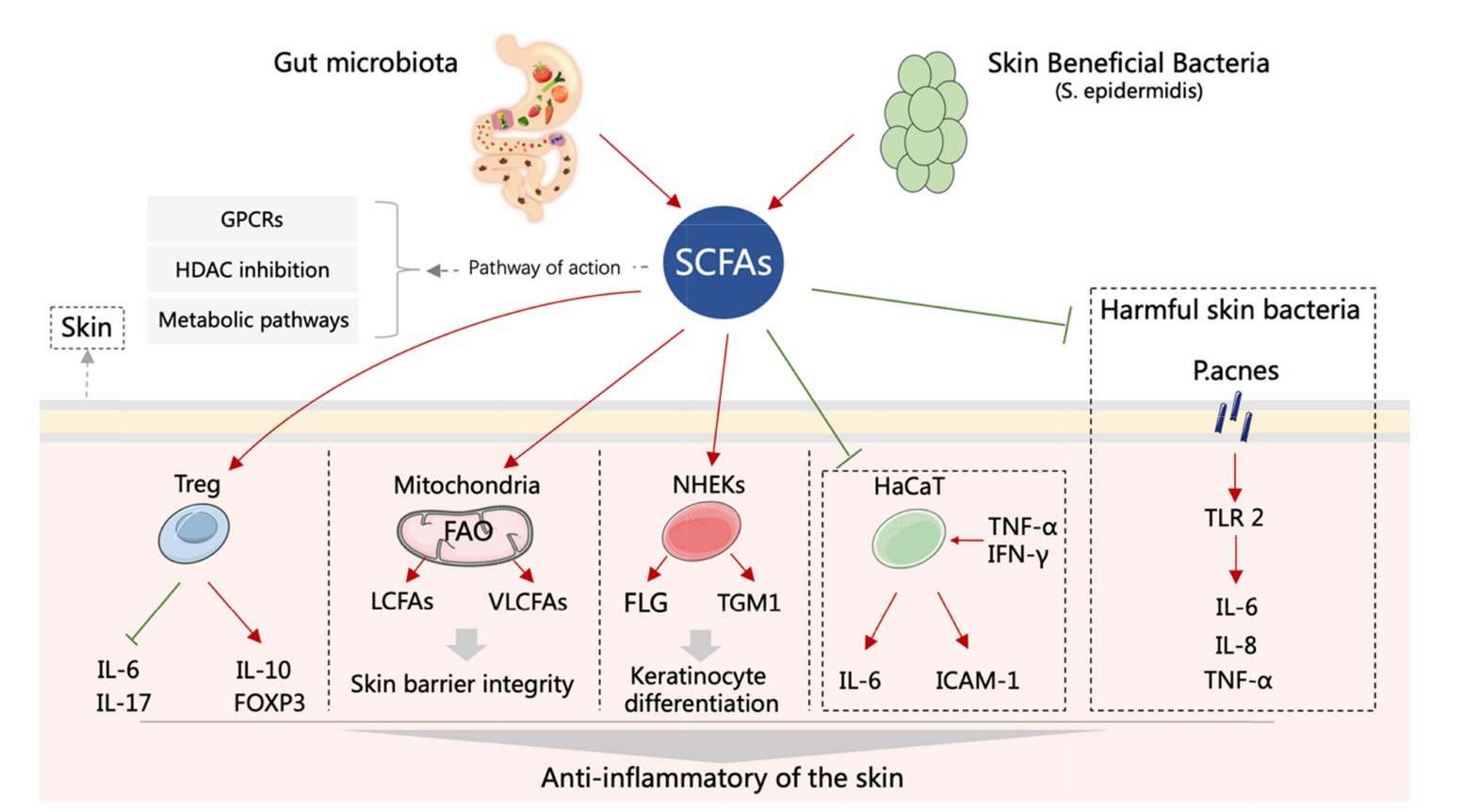 Schematic representation of the roles of SCFAs in inflammatory skin diseases (Xiao et al., 2023)
Schematic representation of the roles of SCFAs in inflammatory skin diseases (Xiao et al., 2023)
In a cohort study of inflammatory bowel disease (IBD), methylation detection of patients' colon biopsy samples using Human Methylome Panel, combined with 5hmC-Seal Seq analysis of hydroxymethylation dynamics, successfully identified that aberrant hypomethylation of the TLR4 gene could be used as an early warning marker of dysbiosis.
Dietary Factors: Folate Deficiency and Global Hypomethylation
Folate deficiency has been demonstrated to result in a reduction in S-adenosylmethionine (SAM) synthesis, thereby triggering a decrease in DNA methylation transferase (DNMTs) activity. An analysis of cord blood from the offspring of mothers with insufficient folate intake during pregnancy by Reduced Representation Bisulfite Sequencing (RRBS) revealed a 2.3-fold increased risk of methylation loss in imprinted control regions (ICRs). To quantify this risk, the research team developed a methylation site tracking protocol based on Bisulfite Sequencing PCR, which, in combination with Targeted DNA Methylation Analysis, detects the methylation status of 50 key sites in plasma, providing an early warning of the risk of neurodevelopmental disorders. To address the limitation of samples in folate metabolism studies, EM-seq technology was used to perform genome-wide methylation analysis of trace fetal free DNA. This method exhibited a 5-fold increase in sensitivity compared to traditional WGBS. It has been applied to a cohort study of 3,000 pregnant women.
Service you may intersted in
Learn More:
The analysis of the microbiome-epigenetic interaction network necessitates the integration of multidimensional technology across the spectrum from population to single cell and from DNA to RNA. The combined application of macro-genome sequencing and ATAC-Seq can localize chromatin accessibility changes induced by exposure to specific strains of bacteria, while scATAC-Seq reveals heterogeneous responses of intestinal epithelial cells to signals from bacterial communities. These technological breakthroughs provide a molecular map for precise intervention in environmentally related diseases.
Multi-Omics Approaches: Bridging Microbial Taxa and Epigenetic Hotspots
Utilizing the Epigenomic Peak Calling and Annotation technique, researchers identified a specific increase in H3K27ac modification within the enhancer region of the PPAR-γ gene in adipose tissue following Akkermansia enrichment. Subsequent application of Hi-C technology enabled the construction of a three-dimensional genomic interaction network, thereby confirming the spatial co-localization of this region with mitochondrial metabolic gene clusters. This multi-omics integration strategy successfully predicted the molecular pathway of high-fat diet in regulating energy metabolism through the colony-epigenetic axis in an obesity model.
In a study of early liver cancer screening, the combination of cfDNA methylation analysis and 5hmC Selective Chemical Labelling Sequencing revealed abnormally elevated 5hmC levels of APC genes in the plasma of patients with intestinal flora disorders. This combination exhibited a diagnostic specificity that was 22% higher than that of traditional tumor markers.
scATAC-Seq: Mapping Chromatin Accessibility in Microbiota-Exposed Tissues
The application of scATAC-seq to intestinal epithelial cells revealed that Lactobacillus (Lactobacillus) colonization significantly increased the openness of the Lgr5 locus's chromatin. Through Epigenomic Differential Peak Analysis, researchers ascertained that specific Clostridium (Clostridium) strains were able to restore accessibility to the Occludin enhancer region in a model of colitis. Utilizing long read sequencing DNA methylation, the study unequivocally demonstrates for the first time that microbial metabolites can directly induce remodeling of the chromatin ring structure.
Addressing the challenge posed by the high noise in single-cell epigenomic data, a noise reduction algorithm was developed. This algorithm, based on Epigenomic Exploratory Analysis, enhanced the detection rate of chromatin opening signals in rare cell subpopulations by 40%.
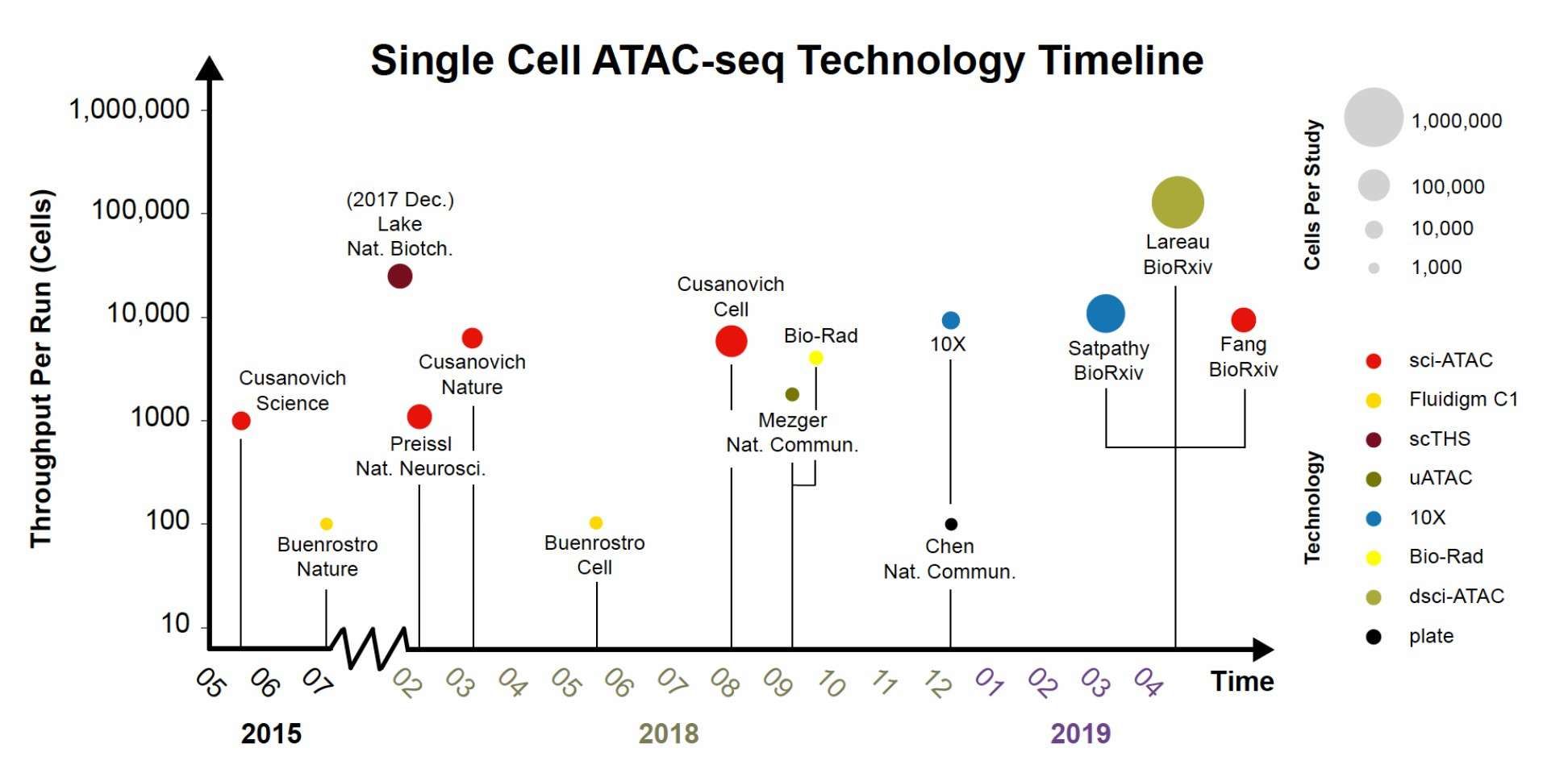 Single Cell ATAC-seg Technology Timeline (Fang, 2019)
Single Cell ATAC-seg Technology Timeline (Fang, 2019)
The phenomenon of environmentally driven epigenetic reprogramming has been demonstrated to be associated with the onset of diseases, while concurrently exhibiting promise in the realm of sustainable agricultural development. This interplay is exemplified by the use of EpiTYPER DNA Methylation Analysis, which unveils epigenetic memory associated with autoimmune diseases. Furthermore, CRISPR-dCas9-mediated targeted methylation has been employed for the purpose of breeding resilient crops, thus underscoring the versatility of these methodologies. These diverse applications serve to transform conventional intervention strategies.
Dysbiosis and Autoimmune Diseases: The Epigenetic Memory of Inflammation
In patients diagnosed with rheumatoid arthritis (RA), a recent study has revealed a correlation between demethylation of the IL-17A motif in synoviocytes and overproliferation of Prevotella. This finding was achieved through the implementation of a Human DNA Methylation Microarray assay. Concurrently, an investigation involving the methylomic analysis of patient stool samples via MethylRAD-Seq methodically identified 12 strain-specific methylases that have emerged as promising therapeutic targets. Presently, a Bisulfite Sequencing PCR-based methylation dynamic monitoring protocol has been employed to assess the effect of methotrexate on the apparent age reversal of RA.
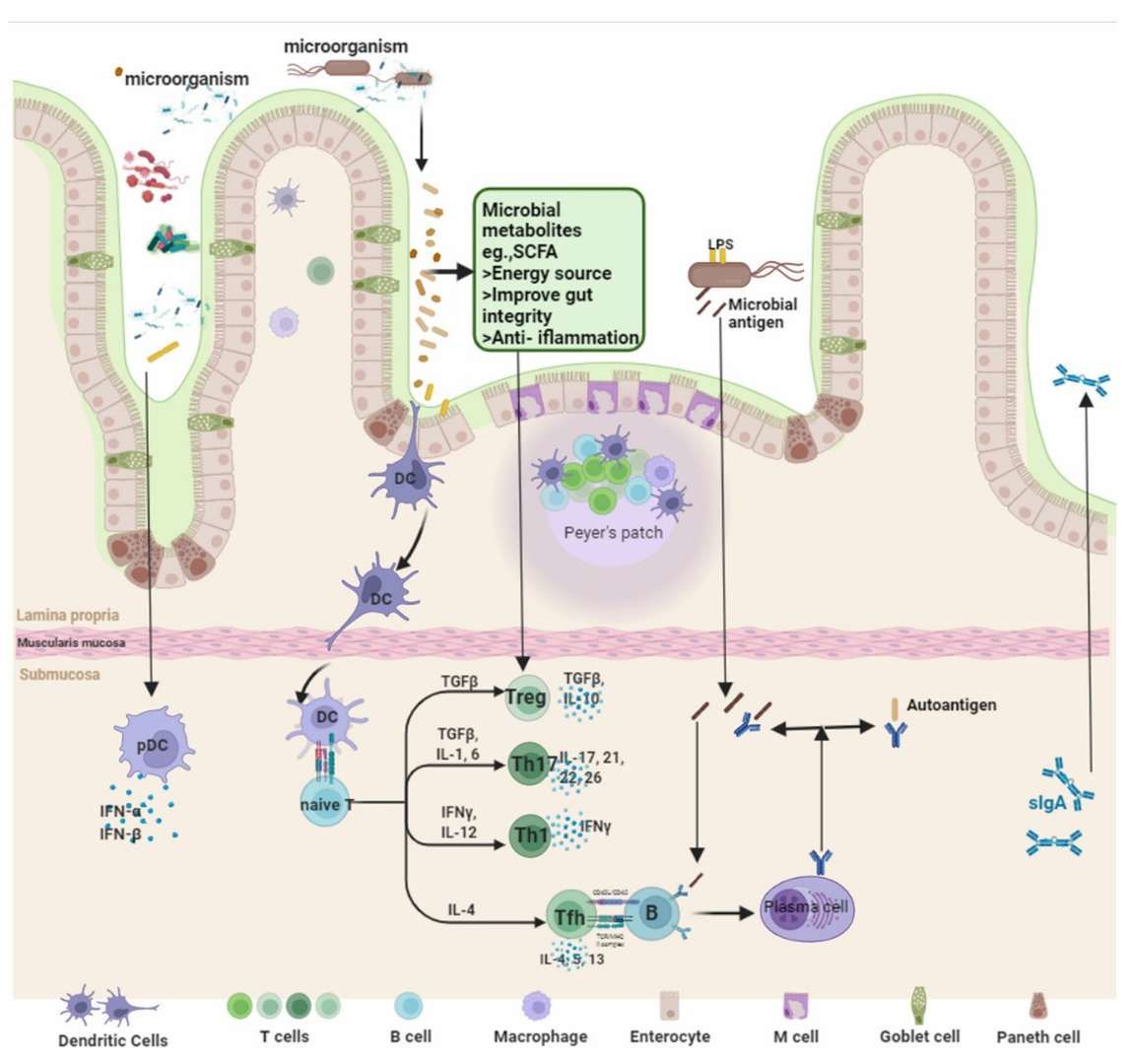 The intervention mechanism of fecal microbiota transplantation (Zeng et al., 2022)
The intervention mechanism of fecal microbiota transplantation (Zeng et al., 2022)
Epigenetic Priming for Stress-Resistant Crops: A New Green Revolution
Utilizing DAP-Seq technology to screen the binding sites of transcription factors associated with salinity resistance in wheat, in conjunction with MRE-Seq to analyze the methylation-sensitive regions, the researchers targeted the silencing of the methylation-sensitive regions of the OsSDIR1 gene by Target Bisulfite Sequencing. Field trial data demonstrated that epigenetically pretreated plants exhibited an 18% yield enhancement under drought stress. The program integrates the Drone-based Epigenomic Monitoring System to validate key epitopes across species by Mouse DNA Methylation Microarray, which reduces the breeding cycle by 40%.
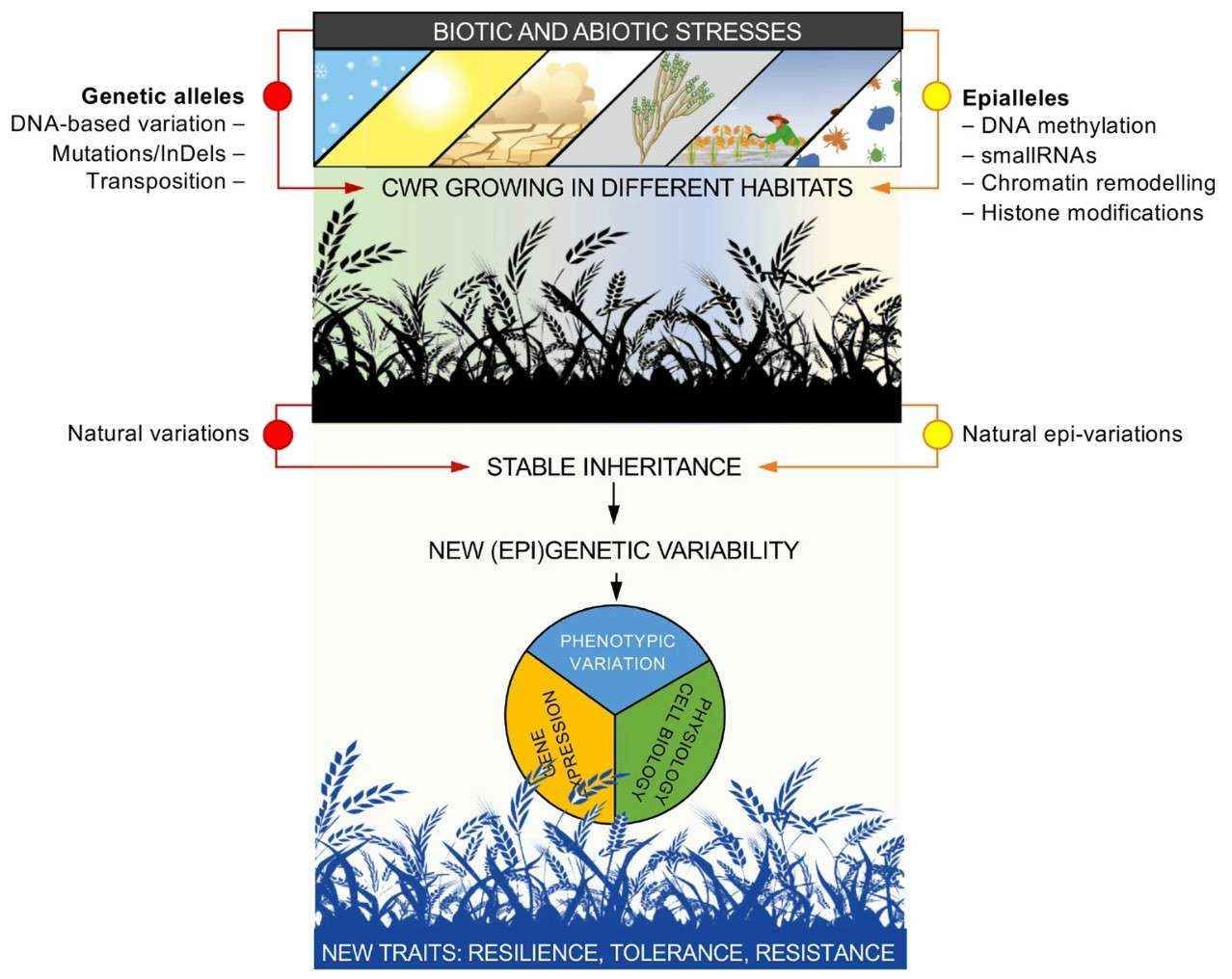 Relationship between epigenetic modifications and crop resistance (Varotto et al., 2022)
Relationship between epigenetic modifications and crop resistance (Varotto et al., 2022)
Despite the potential of environmental epigenetics to provide a novel framework for the management of diseases and the development of agricultural innovations, its advancement is impeded by numerous technical challenges and scientific gaps. First, the spatial and temporal variations in microbiome-host interactions present significant difficulties in the field of detection technology. The brief half-life of gut flora metabolites (e.g., butyric acid persists for only a few minutes) complicates the task of traditional WGBS in capturing their transient methylation effects. Secondly, the complexity of cross-omics data integration underscores the limitations of existing analytical tools. For instance, integrating RNA-seq and epigenomic data analysis remains constrained by batch effects and sample heterogeneity when correlating microbial functional genes with host epigenetic targets. Furthermore, the epigenomic memory of environmental exposures varies across species, and the translation of mouse DNA methylation microarray-based discoveries to crops in agriculture is often invalidated by the low conservation of chromatin three-dimensional structure. A more significant challenge pertains to the sensitivity of cell-free DNA methylation analysis in clinical applications, which is currently limited (e.g., the detection rate of early tumor markers is less than 60%). The tissue-specific resolution of environmental epitopes remains to be achieved, despite the potential of single-cell technology, due to the cost constraints.
In order to overcome the aforementioned challenges, it is imperative to leverage technological innovation and interdisciplinary collaboration. From a technical standpoint, the integration of Long Read Sequencing, DNA Methylation, and ONT Direct RNA Sequencing enables the concurrent analysis of DNA methylation, RNA modification, and microbial transcriptional activity. This approach unveils the intricate epigenomic regulatory network influenced by environmental exposure. Through the optimization of the Epigenomic Exploratory Analysis algorithm, we have the capacity to extract epigenetic response signals from rare cell subpopulations from noisy single-cell data (e.g., scATAC-Seq), achieving a sensitivity of up to 85%. At the application level, it is imperative to establish "environmental epibanking," which involves the longitudinal monitoring of millions of individuals using the Human Methylome Panel and 5hmC-Seal Seq. In the agricultural sector, the development of a DAP-Microbiome-Seal Seq is imperative for longitudinal monitoring of millions of individuals. Furthermore, the creation of a DAP-Seq-driven intelligent breeding platform is crucial for the screening of stress-resistant epigenetic markers by MRE-Seq. This platform can be integrated with Drone-based Epigenomic Monitoring to facilitate real-time phenotype prediction of field crops.
References
Terms & Conditions Privacy Policy Copyright © CD Genomics. All rights reserved.