
In the rapidly evolving field of epigenetics, DNA methylation plays a crucial role in gene regulation and various biological processes. Traditional sequencing technologies often face challenges in accurately detecting DNA methylation patterns due to limitations in read length and resolution. However, with advancements in long-read sequencing technologies, such as PacBio and Oxford Nanopore sequencing, it has become possible to obtain high-quality, long-range sequencing data, enabling comprehensive analysis of DNA methylation patterns.
CD Genomics, a leading genomics service provider with years of experience, offers state-of-the-art Long Read Sequencing DNA Methylation Service to cater to the growing demands of researchers in the field.
Long Read Sequencing Technology for DNA Methylation Detection
Long-read sequencing technologies have revolutionized the study of DNA methylation by providing enhanced capabilities for accurate detection and characterization of methylation patterns. These technologies, including PacBio and Oxford Nanopore sequencing, generate longer sequencing reads, enabling the identification of methylation patterns across larger genomic regions, including repetitive elements and structural variations. This long-read sequencing technology is particularly valuable for studying complex biological phenomena influenced by DNA methylation, such as gene regulation, development, and disease progression.
What Can Our Service Provide?
CD Genomics' Long Read Sequencing DNA Methylation Service offers a comprehensive solution for researchers aiming to explore DNA methylation patterns with high accuracy and resolution.
Advanced Long Read Sequencing Platforms
At CD Genomics, we utilize cutting-edge long-read sequencing platforms, such as PacBio Sequel and Oxford Nanopore MinION/GridION, known for their ability to generate long sequencing reads. These platforms enable us to capture DNA methylation patterns across the genome with unparalleled precision and coverage.
Comprehensive Library Preparation and Sequencing
Our experienced team at CD Genomics employs optimized protocols for library preparation, including DNA fragmentation, end-repair, adapter ligation, and size selection, to ensure high-quality sequencing libraries. Subsequently, we perform sequencing using the selected long-read platforms, generating data that spans kilobases of DNA.
High-Quality Data Analysis
CD Genomics provides robust bioinformatics analysis tailored to extract valuable insights from the generated sequencing data. Our skilled bioinformatics team utilizes state-of-the-art algorithms and pipelines to ensure accurate identification of DNA methylation patterns, differential methylation analysis, annotation of methylation sites, and integration with other omics data for comprehensive interpretation.
Customized Solutions and Flexible Service Options
We understand that research goals can vary, and therefore, we offer customizable service options to meet specific research requirements. Whether it's whole-genome methylation analysis, targeted region analysis, or integration with other data types, our service can be tailored to suit your needs.
Service Advantages
Choosing CD Genomics' Long Read Sequencing DNA Methylation Service provides researchers with numerous advantages:
Single-Nucleotide Resolution Detection
Our service offers single-nucleotide resolution detection of DNA methylation, enabling precise identification and characterization of methylation patterns at the base level. This level of resolution is crucial for understanding the functional implications of DNA methylation in various biological contexts.
High Specificity and Sensitivity
We employ m1A-specific antibodies or other specific enrichment methods to ensure high specificity and sensitivity in capturing methylated fragments during sequencing. This targeted enrichment approach enhances the accuracy and reliability of DNA methylation analysis.
One-Stop Service
CD Genomics provides a streamlined one-stop service, starting from sample submission to comprehensive data analysis. Customers only need to provide their cells, tissues, body fluids, or total RNA, and we take care of the entire service process, including enrichment, library preparation, sequencing, and bioinformatics analysis.
Professional Bioinformatics Analysis
Our dedicated bioinformatics team possesses extensive expertise in analyzing high-throughput sequencing data. We offer comprehensive and customized data analysis solutions, including quality control assessment, identification of methylation enrichment regions, differential methylation analysis, gene expression analysis, and integration of methylation data with other genomic features.
Service Workflow
Data Analysis Content
The CD Genomics Long Read Sequencing DNA Methylation Service offers extensive data analysis content, including but not limited to:
| Data Analysis Content | Description |
|---|---|
| Quality control and assessment | - Data quality control - Alignment statistics - Insert fragment length statistics - Whole-genome coverage statistics - Sample saturation curve |
| Identification and statistics of methylation enrichment regions | - Identification, annotation, and basic statistics of methylation enrichment regions - Distribution on chromosomes and genomic elements |
| Analysis of differentially modified methylation sites | - Peak overlap analysis - Identification and annotation of differentially modified methylation sites - Distribution on chromosomes and genomic elements |
| Expression level analysis | - Identification of lncRNAs and circRNAs - Analysis of gene expression levels - Clustering analysis - Correlation analysis |
| Analysis of differential methylation modifications and associated genes | - Identification of differentially expressed genes - Functional enrichment analysis - Analysis of the association between differential methylation modifications and genes |
Long-read whole-genome methylation patterning using enzymatic base conversion and nanopore sequencing
Abstract
This case study explores the research presented in the article "Long-read whole-genome methylation patterning using enzymatic base conversion and nanopore sequencing" published in Nucleic Acids Research. The study focuses on unraveling the complex landscape of DNA methylation using long-read sequencing technologies. By employing enzymatic base conversion and nanopore sequencing, the authors provide valuable insights into the high-resolution methylation patterns across the entire genome. The case study aims to provide a comprehensive overview of the research, including the methodologies used, key findings, and their implications for understanding the epigenetic regulation of the genome.
Methods
The researchers employed enzymatic base conversion techniques, such as TET-assisted pyridine borane sequencing (TAPS), to achieve site-specific conversion of methylated cytosines to uracils. The converted DNA was then subjected to nanopore sequencing, which allows for long-read sequencing of DNA molecules. The authors utilized specialized bioinformatics pipelines to analyze the raw nanopore sequencing data and determine the methylation patterns across the genome.
Results
The study successfully demonstrated the feasibility of long-read whole-genome methylation patterning using enzymatic base conversion and nanopore sequencing. The researchers identified and characterized methylated cytosines at single-base resolution, providing a comprehensive map of DNA methylation across the genome. They observed distinct methylation patterns in different genomic regions, including gene promoters, enhancers, and repetitive elements. Moreover, the study revealed the presence of methylation heterogeneity and allele-specific methylation patterns, emphasizing the dynamic nature of DNA methylation.
For example, Long-read methylation analysis would determine the methylation patterns on long single DNA molecules. By the obtained long read data, allele-biased methylation patterns will be more clearly detected. In fact, by the long-read methylation analysis using nanoEM methods, we could dissect the methylation pattern of imprinting genes and structural variations in an allele-sensitive manner (Figures 1C–E and 5G). We were also able to detect the methylation within repetitive sequences (Figure 2F).
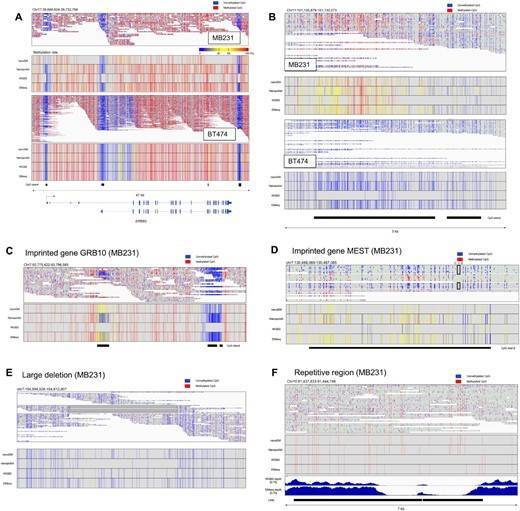 Figure 1 Methylation patterns of several genes and regions.
Figure 1 Methylation patterns of several genes and regions.
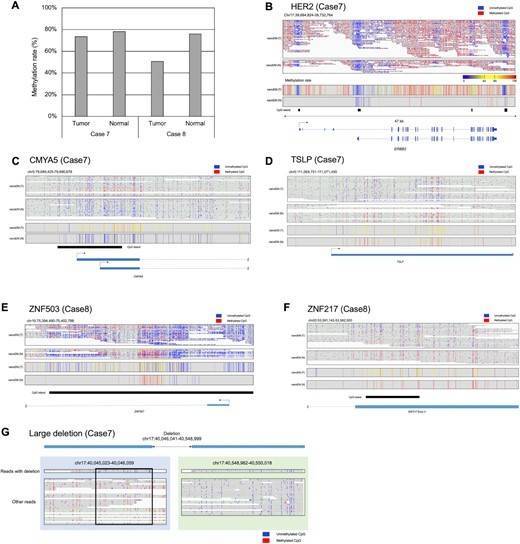 Figure 2 Applications to clinical samples.
Figure 2 Applications to clinical samples.
Conclusion
The research on long-read whole-genome methylation patterning using enzymatic base conversion and nanopore sequencing provides valuable insights into the dynamic landscape of DNA methylation. This case study highlights the key methodologies and findings, showcasing the power of combining enzymatic base conversion with nanopore sequencing to achieve comprehensive methylation profiling. The insights gained from this study pave the way for further advancements in our understanding of the epigenetic regulation of the genome and its implications in various biological processes and human health. Future studies could further investigate the functional consequences of specific methylation patterns, explore the interplay between DNA methylation and other epigenetic modifications, and examine the potential of DNA methylation patterns as biomarkers for disease diagnosis and prognosis.