
EM-seq (Enzymatic Methyl Sequencing) Introduction
Over the decades of human genome sequencing, researchers have established connections between genetic factors and many diseases. However, the nucleotides encoding the genomic DNA of protein-coding genes are only a fraction of the genetic factors that influence cellular function and overall health. Epigenetics, which refers to heritable changes that occur in certain circumstances without altering the DNA sequence, also plays a crucial role. DNA methylation, a well-studied epigenetic mark, involves the chemical modification of cytosine and adenine residues. Cytosine methylation commonly occurs at CG sequences in the genome, known as CpG sites, and it extensively regulates gene expression in a cell-specific manner. Genome-wide association studies (GWAS) have linked changes in DNA methylation to complex diseases such as cancer and obesity, as well as complex biological states like aging and development. In recent years, methylation sequencing has expanded beyond gene expression and has gained attention in disease diagnostics. Recent studies have demonstrated that differential methylation serves as an informative and sensitive marker in cancer detection, regardless of its correlation with gene expression.
Currently, there have been numerous studies on DNA methylation in cancer liquid biopsies, such as cfDNA, and it has received increasing attention. Conventional methods for DNA methylation studies are based on harsh chemical transformation using bisulfite, which causes significant DNA damage and requires high DNA input. However, the content of cfDNA in blood is usually low, typically in the range of a few tens of nanograms, making it challenging to meet the requirements of conventional bisulfite sequencing methods.
EM-seq Service
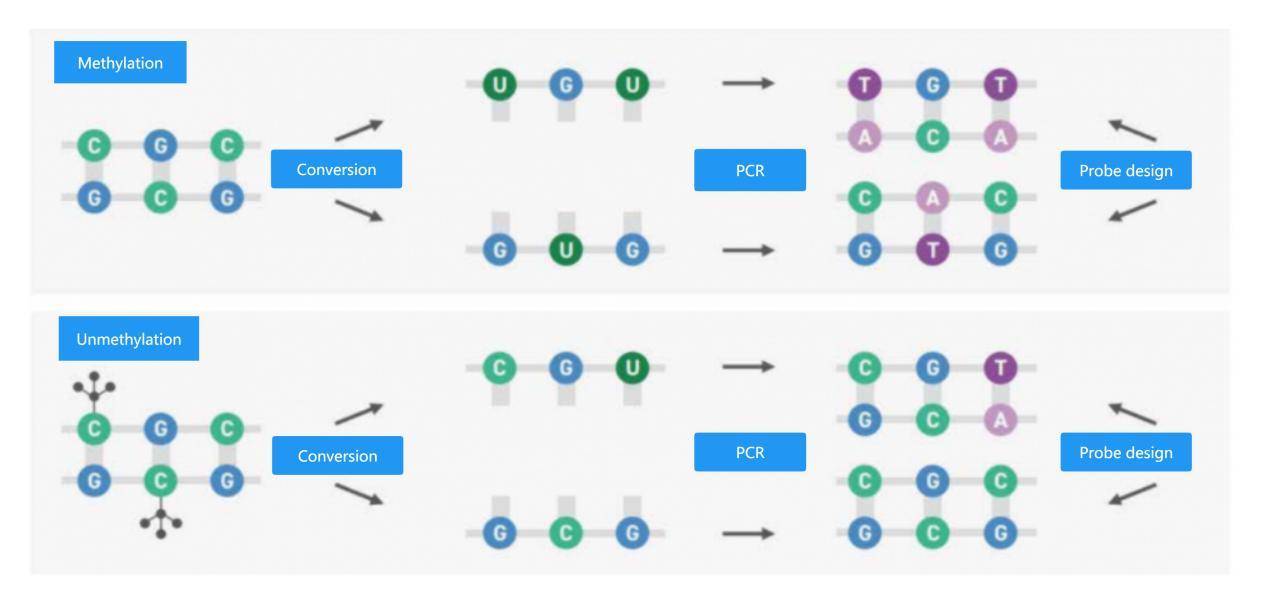
Therefore, CD Genomics has introduced EM-seq (Enzymatic methyl sequencing), a technology based on enzymatic conversion that enables single-base resolution of DNA methylation sites. It is suitable for biomarker research in cancer early screening liquid biopsy samples, such as plasma cfDNA, urine cfDNA, cerebrospinal fluid cfDNA, as well as the study of cell differentiation using minute amounts of individual cells, such as oocytes, sperm cells, embryos, and so on. EM-seq provides a robust and complete solution for identifying methylated regions in the human genome. During the library preparation process, a unique enzymatic conversion method is employed, which causes much less damage to DNA and requires less sample input, resulting in higher quality and better-performing libraries. The custom-designed Twist methylation probe panel provides efficient and specific probes for CpG detection, enabling targeted enrichment. Optimized hybridization reagents increase workflow flexibility and improve on-target rates. Methylation sequencing involves enzymatic or chemical methods that allow unmethylated cytosine to undergo a series of reactions to convert into uracil while methylated cytosine remains intact (Figure 1). During amplification, thymine is added complementary to the uracil on the opposite strand, resulting in the introduction of a cytosine in the original position of the unmethylated cytosine. As shown in the diagram, the final products of the sequence are asymmetric, forming two different double-stranded DNA molecules after conversion (top panel), while the same process produces different sequences for methylated DNA (bottom panel).
EM-seq Advantages
Enzymatic conversion minimizes DNA damage, allowing for ultra-low DNA input requirements as low as 10ng.
Excellent alignment rate and GC uniformity.
High sensitivity with single-base resolution.
Able to detect an additional 15% of methylated sites compared to bisulfite-based methods.
Flexibility to target different regions of interest.
For human (hg38), the designed length is 134Mb, covering approximately 4 million CpG sites. The design is based on new databases such as UCSC, Ensembl, ENCODE, and focuses on regions where known methylation can impact gene regulation: 57% CpG open seas (interCGI), 21% CpG island shores, 15% CpG islands, and 8% CpG island shelves.
EM-seq Workflow
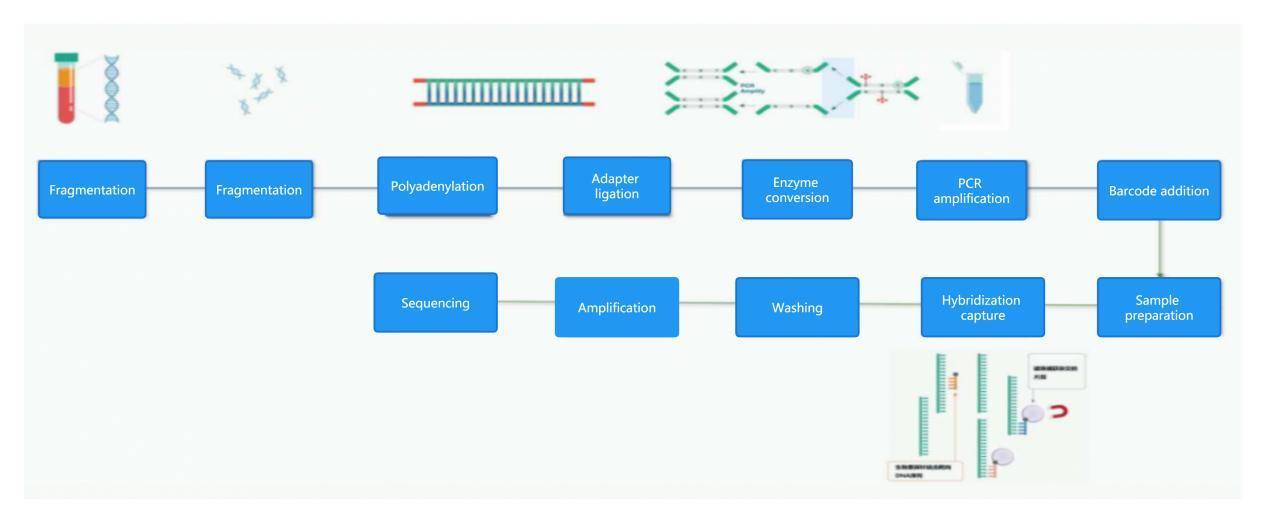
Technology Principles
During the first round of reactions, ten-eleven translocation 2 (TET2) enzymes convert methylated cytosine (5mC and 5hmC) into 5-carboxyl cytosine (5caC) and oxidized enhancer glucosyl acid (5ghmC). These reactions protect 5mC and 5hmC from downstream deamination. Subsequently, APOBEC causes DNA denaturation before deaminating cytosine to uracil. The following polymerase chain reaction (PCR) amplification converts modified 5mC or 5hmC back to cytosine and converts uracil to thymine. After PCR, the nucleotide sequence is identical to that obtained from bisulfite conversion, making EM-seq compatible with existing analysis workflows such as Bismark and bwa-meth.
In both EM-seq and BS-seq, enzymes and sodium bisulfite are used to convert unmethylated cytosines to thymines, and the conversion efficiency is similar. CpG-methylated pUC19 DNA and unmethylated Lambda DNA are chosen as quality control objects for library conversion. The results show that both library conversion methods achieve a conversion rate of 99.5%, measured as the percentage of cytosines converted to thymines at non-CpG sites.
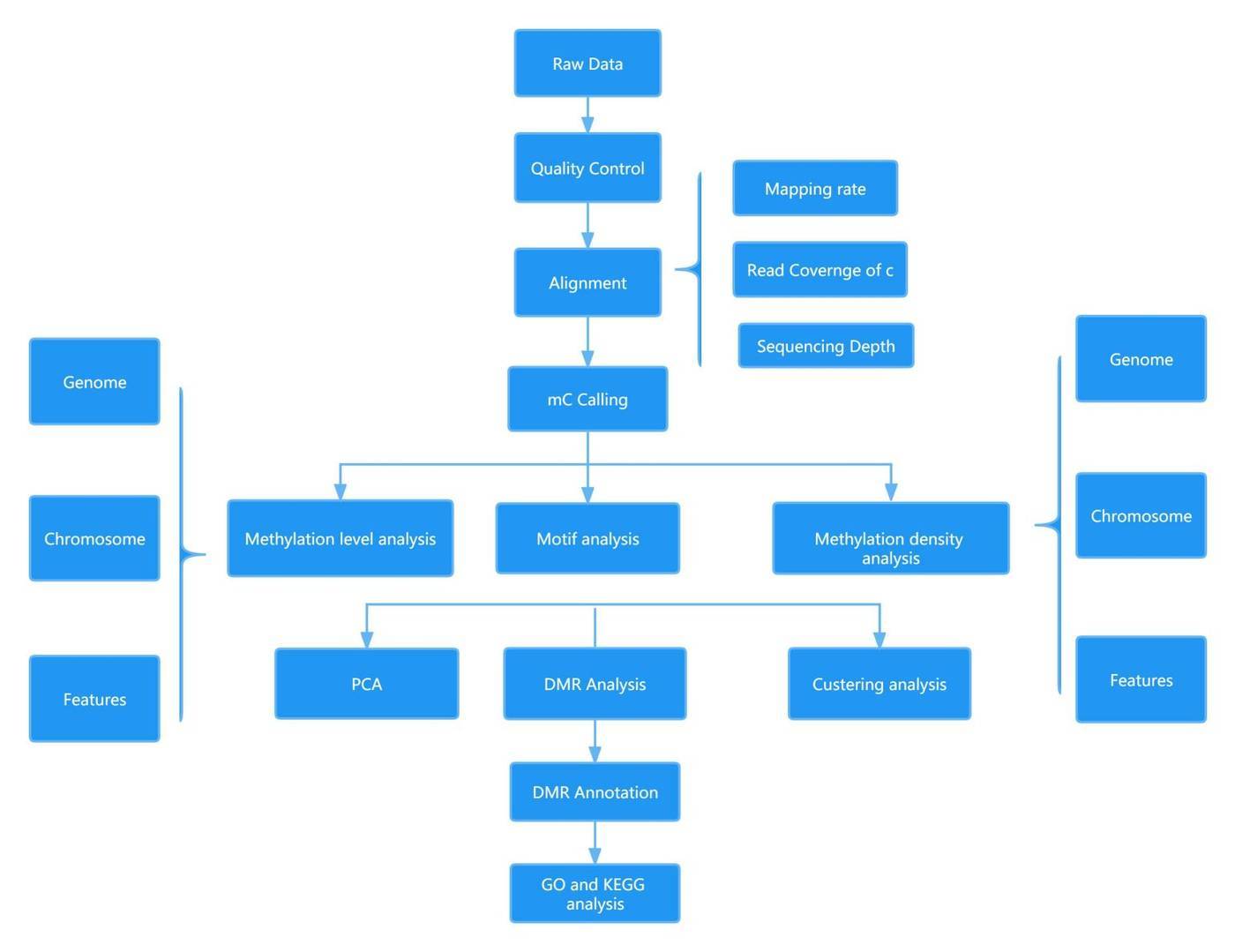
EM-seq Data Analysis
Sample requirements
Sample Type: Human, particularly suitable for liquid biopsy samples such as plasma, urine, cerebrospinal fluid, saliva, etc. It is also applicable to FFPE samples, which are low-volume samples.
Sample Collection: Due to the low quantity, short half-life, and poor stability of cfDNA released by tumors, it is recommended to use dedicated cfDNA collection tubes (10ml) containing fixatives for sample collection to stabilize cfDNA. Store at 4°C and separate plasma within 48 hours.
Long-term storage at -80°C.
Total Sample Amount: Each sample should have a minimum DNA starting amount of 10ng.
Sample Concentration: Use Qubit for accurate quantification of DNA concentration.