Overview of WGBS: Principle, Workflow, Application and Development
Epigenetic modifications, particularly DNA methylation, play an indispensable role in modulating gene expression, cell differentiation, embryogenesis, and disease pathogenesis. WGBS technology has transformed the field of epigenetics by enabling genome-wide, high-resolution mapping of DNA methylation patterns, thereby facilitating in-depth investigations into the molecular mechanisms underlying these biological phenomena.
The Principle of WGBS
The foundation of WGBS lies in the chemical conversion of DNA by bisulfite. During this process, unmethylated cytosine (C) is deaminated to uracil (U), while methylated cytosine (5-methylcytosine, 5mC) remains unaltered due to the protection of the methyl group. Subsequent PCR amplification transmutes U to thymine (T), allowing for the discrimination of methylated and unmethylated C sites based on the sequencing data. To prepare the bisulfite-treated DNA fragments for high-throughput sequencing, library construction procedures such as end repair, addition of A-tail, and ligation of sequencing adaptors are essential. These steps ensure the fragments are compatible with the sequencing platform and primers.
The end of DNA fragments transformed by bisulfite usually suffers from damage and the sequence composition changes. In order to carry out high-throughput sequencing, it is necessary to construct a sequencing library. Library construction generally includes terminal repair, adding A tail, connecting sequencing linker and other steps. Through these operations, the two ends of the transformed DNA fragment have suitable binding sites of sequencing primers and the structure of high-throughput sequencing platform, so that sequencing can be carried out on the sequencer.
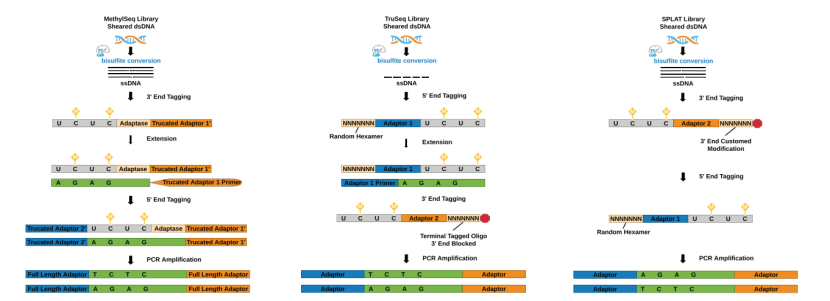 Principle in WGBS analysis (Gong et al., 2022)
Principle in WGBS analysis (Gong et al., 2022)
The Workflow of WGBS
The WGBS workflow commences with the extraction of high-quality genomic DNA from biological specimens using optimized methods like phenol-chloroform extraction or silica gel column adsorption, tailored to the sample type. The extracted DNA then undergoes bisulfite treatment, where precise control of reaction parameters (temperature, time, and bisulfite concentration) is critical to achieve efficient conversion and maintain DNA integrity. Library construction involves flattening the DNA fragment ends, appending an A-tail, and ligating sequencing adaptors, followed by PCR amplification to enrich the library. Subsequently, the library is sequenced on a high-throughput platform (e.g., Illumina), with sequencing depth and coverage being key determinants of data quality.
DNA extraction: Extracting high-quality genomic DNA from biological samples (such as tissues and cells) is the first step of WGBS experiment. Appropriate methods should be adopted in the extraction process to ensure the integrity and purity of DNA and avoid DNA degradation and pollution. Commonly used DNA extraction methods include phenol-chloroform extraction and silica gel column adsorption, and the corresponding extraction conditions need to be optimized for different types of samples.
Transformation of bisulfite: The extracted genomic DNA was treated with bisulfite to realize the transformation from unmethylated C to U. The key of this step is to control the reaction conditions, such as temperature, time and bisulfite concentration, to ensure the transformation efficiency and DNA integrity.
Library construction: The transformed DNA was used for library construction. Firstly, the end of DNA fragment is repaired to make it flat-ended; Then a tail is added to the 3' end of the fragment, which is convenient to connect with the sequencing linker with a t tail; After connecting sequencing adapters, the library fragments were amplified and enriched by PCR.
High-throughput sequencing: The constructed library was sequenced on high-throughput sequencing platform (such as Illumina sequencing platform). In the process of sequencing, the sequencer sequences the DNA fragments of the library base by base according to the principle of base complementary pairing, and generates massive sequencing data. Sequencing depth and coverage are important indicators to measure the quality of sequencing data. Higher sequencing depth can improve the accuracy of methylation site detection, while sufficient coverage can ensure that methylation information in the whole genome is fully captured.
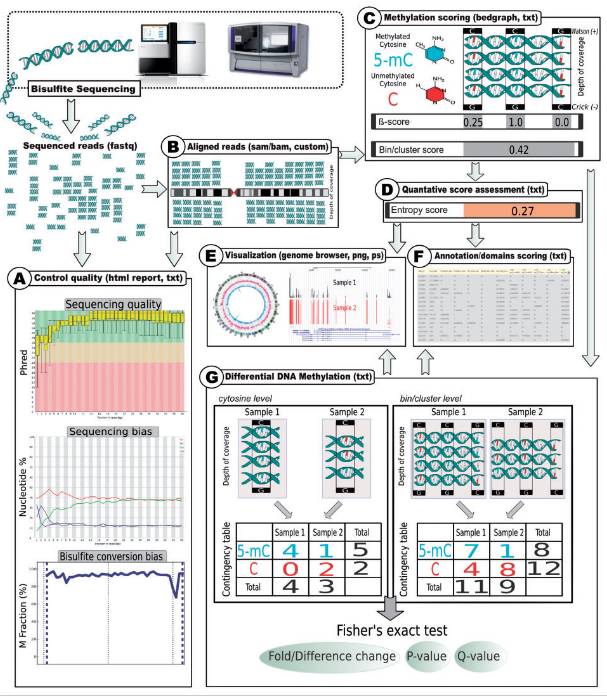 Workflow in genome-wide methylation analysis (Adusumalli et al., 2014)
Workflow in genome-wide methylation analysis (Adusumalli et al., 2014)
Service you may intersted in
How to Analyse WGBS Data
Data analysis in WGBS is a multi-step process. Initially, raw sequencing data is preprocessed to eliminate linker sequences, low-quality reads, and reads with high N ratios, enhancing data reliability. Alignment of the preprocessed data to the reference genome is accomplished using specialized tools (e.g., Bismark) that account for the bisulfite-induced sequence changes. Methylation sites are then identified by analyzing the aligned reads, considering factors like sequencing depth and base quality to ensure accuracy. Methylation levels are calculated as the ratio of methylated cytosine to total cytosine at each locus. Differential methylation analysis between samples or groups is performed to identify regions with potential regulatory significance. Finally, data visualization tools (e.g., Integrative Genomics Viewer, ggplot2 in R) are employed to present methylation data in an intuitive graphical format, facilitating data interpretation.
Data preprocessing: The original sequencing data contains a large number of linker sequences, low-quality bases and other impurities, so it needs to be preprocessed. It mainly includes removing linker sequence, filtering low-quality reads (such as reads with high base ratio whose mass value is lower than the set threshold) and removing reads with high N ratio. Through data preprocessing, the data quality can be improved and a reliable data base can be provided for subsequent analysis.
Alignment to reference genome: The preprocessed data need to be compared to the reference genome to determine the position of each read in the genome. Due to the change of DNA sequence after bisulfite transformation, the conventional comparison software can not be directly applied, so it is necessary to use a comparison tool specifically for bisulfite transformation data, such as Bismark. These tools will consider the transformation from C to T in the alignment process, so as to accurately locate sequencing reads on the reference genome.
Methylation site calling: After the comparison is completed, the methylation sites are determined by analyzing the comparison results. Generally, it is judged whether the site C is methylated or not according to whether it is C or T in sequencing reads. In order to improve the accuracy of methylation site judgment, it is necessary to consider factors such as sequencing depth and base mass value, and set appropriate thresholds to filter methylation sites with low reliability. Commonly used methylationlocus Calling tools include methylKit and so on.
Methylation level analysis: Calculate the methylation level of each locus, gene region or specific region of genome. Methylation level is usually expressed as the sum of methylated cytosine number. Cytosine Number (methylation and unmethylation) ratio. By comparing the methylation levels of different samples or different treatment groups, differential methylation regions (DMRs) with potential regulatory effects in biological processes can be mined. Analyzing the gene function and related pathways of DMRs is helpful to reveal the regulation mechanism of DNA methylation in biological process.
Data visualization: Use a variety of visualization tools (such as Integrative Genomics Viewer, IGV, Ggplot2 in R language) will display methylation data in an intuitive graphical way. For example, taking genome coordinates as the horizontal axis and methylation level as the vertical axis, the distribution characteristics of methylation level within the whole genome can be clearly displayed; At the gene level, the relationship between gene structure and methylation level can be drawn, and the methylation status of different regions of the gene (promoter, exon, intron, etc.) can be visualized.
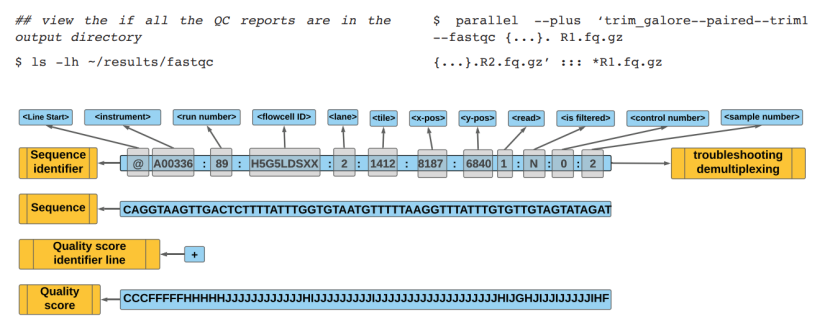 FASTQ file elements (Gong et al., 2022)
FASTQ file elements (Gong et al., 2022)
If you want to learn more about the WGBS, please refer to:
Advantages of WGBS
WGBS offers several distinct advantages. Its single-base resolution empowers researchers to precisely discern methylation status at individual cytosine sites, enabling detailed exploration of the fine-tuning mechanisms of DNA methylation. The genome-wide coverage ensures a comprehensive assessment of methylation patterns across all genomic regions, facilitating the discovery of novel regulatory elements. High detection accuracy, achieved through the combination of bisulfite conversion and high-throughput sequencing, allows for the reliable detection of low-level methylation changes. Moreover, WGBS is applicable to a wide range of sample types, enhancing its versatility in research.
Single base resolution: WGBS can accurately distinguish methylated cytosine from unmethylated cytosine to a single base, which makes it possible to further study the fine regulation mechanism of DNA methylation. This enables researchers to analyze the small differences of methylation patterns in the whole genome, which is of great significance to reveal the key regulatory sites in complex biological processes.
Whole genome coverage: This technique can analyze the methylation of the whole genome, covering all gene regions without preference, including promoters, coding regions, non-coding regions and intergenic regions. Compared with other methylation detection techniques only targeting specific regions, WGBS can comprehensively and systematically describe the methylation landscape of genome, which is helpful to discover new methylation regulatory elements and potential regulatory mechanisms.
High accuracy: Through the combination of bisulfite conversion and high-throughput sequencing, WGBS has high accuracy in detecting methylation sites. Under the strict control of experimental conditions and data analysis flow, low-level methylation modification can be reliably detected, which provides a strong guarantee for studying the subtle changes of DNA methylation in the process of disease occurrence and development.
The Disadvantages of WGBS
Despite its merits, WGBS has notable limitations. The experimental cost is substantial, primarily due to the complexity of the procedure involving multiple steps and the requirement for high sequencing depth. The bisulfite treatment can cause DNA damage and incomplete conversion, potentially introducing errors in methylation site identification. Data analysis is highly complex, demanding advanced bioinformatics knowledge and significant computational resources to handle the vast amount of data and the unique challenges posed by bisulfite-altered sequences. Additionally, strict sample quality requirements may limit its application in certain scenarios.
The experimental cost is high: WGBS experiment involves a number of complicated steps, including DNA extraction, bisulfite transformation, library construction and high-throughput sequencing, etc. Each step requires the use of specific reagents and equipment, and requires high sequencing depth, resulting in relatively high experimental cost. This limits the application of this technology in large-scale sample research to some extent.
Complex data processing: The generated marine sequence data need to be analyzed by complex pretreatment, comparison and methylation site calling, which requires high computing resources and professional skills of analysts. In addition, due to the change of DNA sequence caused by bisulfite transformation, conventional bioinformatics analysis tools can not be directly applied, and special software and algorithms are needed, which increases the difficulty and complexity of data analysis.
DNA damage and incomplete transformation: The process of bisulfite treatment will cause some damage to DNA, which may lead to DNA fragmentation and information loss. At the same time, it is difficult to achieve 100% transformation efficiency in the transformation process, and some unmethylated C is not completely transformed into U, which may affect the accuracy of methylation site judgment and need to be considered and corrected in experimental design and data analysis.
New Types of WGBS
The new WGBS is optimized on the basis of tradition, and the sample processing is improved to improve the transformation efficiency and DNA integrity and reduce the degradation risk. Innovate the strategy of database construction, improve the efficiency and uniformity of database construction, and reduce the preference. In the sequencing and analysis process, with the help of new platforms and algorithms, more Qualcomm quantity and more accurate interpretation can be achieved, such as optimizing methylation site identification by using machine learning. These improvements have reduced the cost, improved the sensitivity and resolution, and promoted the development of epigenetics by applying them in the fields of mechanism analysis of complex diseases and dynamic research of embryo development.
PBAT-WGBS
Principle: this is a method of transforming first and then building a library. First, the sample DNA is treated with sulfite to convert unmethylated C bases into U bases, and then the linker labeling and library construction are carried out. Through two rounds of random primer amplification, the sulfite-treated samples were connected with complete connectors to form a complete library.
Advantages: WGBS analysis can be carried out with only a small amount of DNA, which avoids the problem of sequencing template breakage caused by bisulfite, and omitting DNA connection and gel purification steps is also helpful to improve efficiency.
T-WGBS
Principle: By adding a specific tag sequence to the DNA fragment, and then carrying out sulfite treatment and high-throughput sequencing. In the process of sequencing, different DNA fragments are distinguished by identifying tag sequences, thus improving the efficiency and accuracy of sequencing.
Advantages: it can improve the efficiency and accuracy of library construction, reduce the deviation and error in the process of library construction, and is suitable for large-scale methylation research and clinical diagnosis.
Single cell WGBS
Principle: Combining single cell isolation technology and WGBS technology, the whole genome methylation of a single cell was sequenced and analyzed. Usually, it is necessary to isolate single cells first, then amplify the whole genome, and then carry out sulfite treatment and high-throughput sequencing.
Advantages: It can reveal the heterogeneity of methylation between cells for research. cell differentiation Epigenetic regulation mechanism in the process of development and disease occurrence provides more detailed information.
Nano-porous WGBS
Principle: DNA molecules are directly sequenced by using nanopore sequencing technology, and DNA methylation status can be detected in real time during the sequencing process. Nanopore sequencing technology is a sequencing technology based on single molecule electrical detection, which determines the DNA sequence and methylation state by detecting the current changes generated by DNA molecules passing through nanopores.
Advantages: sulfite treatment and PCR amplification are not needed, which avoids possible deviations and errors in these processes; It can directly sequence long fragments of DNA, which improves the genome coverage and the detection accuracy of methylation sites; Real-time sequencing and methylation detection can be realized, which provides a faster and more convenient method for studying epigenetic regulation mechanism.
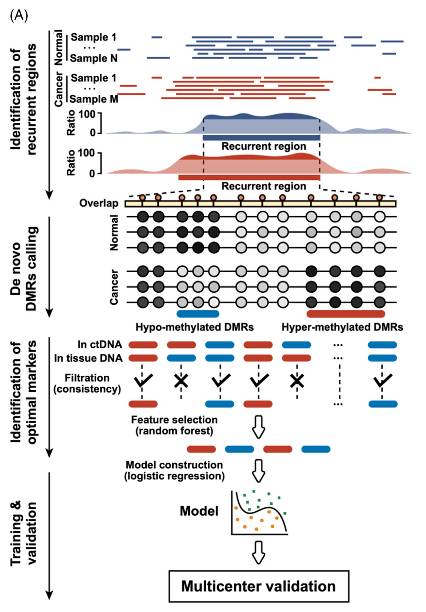 Computational analysis workflow of recurrent regions (Gao et al., 2022)
Computational analysis workflow of recurrent regions (Gao et al., 2022)
If you want to learn more about the WGBS, please refer to:
Application of WGBS
The occurrence and development of tumor is closely related to abnormal DNA methylation. Through the genome-wide methylation analysis of tumor tissues and normal tissues by WGBS, tumor-specific methylation changes can be found. For example, in the study of colorectal cancer, the hypermethylation of promoter regions of several tumor suppressor genes was identified by WGBS technology, and the abnormal methylation of these genes led to their silence, thus promoting the occurrence and development of tumors. At the same time, WGBS can also be used for early diagnosis and prognosis evaluation of tumors, providing potential biomarkers for precise treatment of tumors.
Embryonic development involves precise regulation of gene expression, and DNA methylation plays a key role in it. WGBS analysis of embryos at different developmental stages can reveal the dynamic changes of genome-wide methylation patterns during embryonic development. It was found that the methylation level of genome changed significantly before and after embryo implantation, and the methylation status changes of some key developmental regulatory genes were closely related to the temporal and spatial specificity of gene expression, which provided important clues for further understanding the epigenetic regulatory mechanism of embryo development.
DNA methylation is involved in regulating plant growth and development, flowering and fruiting, and response to environmental stress. Using WGBS technology to analyze methylation of different varieties of plants or plants under different environmental conditions, methylation sites and regions related to plant agronomic traits can be mined. For example, in rice research, some differences in methylation modification of genes related to rice yield, quality and stress resistance were found through WGBS, which provided new targets and theoretical basis for rice molecular breeding.
WGBS has revolutionized the study of DNA methylation, providing a powerful means to explore the epigenetic regulation of biological processes. Despite its existing challenges, continuous technological advancements in sample processing, sequencing, and data analysis are expected to overcome these limitations. The development of novel WGBS variants further expands its potential applications. As WGBS continues to evolve, it will undoubtedly play an increasingly crucial role in advancing our understanding of epigenetics and its implications for human health, agriculture, and developmental biology.
References
- Gong Ting, Heather Borgard, Zhang Zao, Chen Shaoqiu and Deng Youping. "Analysis and Performance Assessment of the Whole Genome Bisulffte Sequencing Data Workffow: Currently Available Tools and a Practical Guide to Advance DNA Methylation Studies." Small Methods (2022) 2101251. https://doi.org/10.1002/smtd.202101251
- Swarnaseetha Adusumalli, Mohd Feroz Mohd Omar, Richie Soong and Touati Benoukraf. "Methodological aspects of whole-genome bisulfite sequencing analysis." Briefings in Bioinformatics (2014) 369-379 doi:10.1093/bib/bbu016
- Gao Yibo, Zhao Hengqiang, Ke An, Liu Zongzhi and Hai Luo. "Whole-genome bisulfite sequencing analysis of circulating tumour DNA for the detection and molecular classification of cancer." Clin. Transl. Med. (2022) e1014 https://doi.org/10.1002/ctm2.1014
- Slivia Gravina, Dong Xiao, Yu Bo and Jan Vijg. Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome." Genome Biology (2016) 17:150. DOI 10.1186/s13059-016-1011-3
- Wang Qi, Gu Lei, Andrew Adey, Wang Wei and Dieter Weichenhan. "Tagmentation-based whole-genome bisulfite sequencing." Nature Protocols (2013): 2022-2032. doi:10.1038/nprot.2013.118
- Fumihito Miura, Yukiko Shibata, Miki Miura and Takashi Ito. "Post-bisulfite Adaptor Tagging Based on an ssDNA Ligatio Technique (tPBAT)." Epigenomics (2023): 2577. https://doi.org/10.1007/978-1-0716-2724-2_2
- Sofia Battaglia, Kevin Dong, Wu Jingyi, Chen Zeyu and Bradley E. Bernstein. "Long-range phasing of dynamic, tissue-specific and allele-specific regulatory elements." Nature Genetics (2022): 1504–1513. https://doi.org/10.1038/s41588-022-01188-8
! For research purposes only, not intended for clinical
diagnosis, treatment, or individual health assessments.



