




We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
In the burgeoning field of epigenetics, DNA methylation has emerged as a pivotal mechanism in the regulation of gene expression, cell differentiation, development, and disease pathogenesis. Reduced Representation Bisulfite Sequencing (RRBS) has garnered significant attention in recent years as an efficient, cost-effective, and high-resolution technique for dissecting DNA methylation patterns.
RRBS is a sophisticated methodology that integrates the precision of restriction endonuclease digestion with the discriminatory power of bisulfite sequencing. By selectively cleaving the genome with specific restriction enzymes, such as MspI, which targets the CCGG sequence, RRBS enriches for CpG-rich regions encompassing promoters and CpG islands. This targeted approach not only streamlines the genomic complexity but also allows for in-depth analysis of these key regulatory domains. Subsequent bisulfite treatment converts unmethylated cytosine residues to uracil, while methylated cytosines remain unaltered. Sequencing of the treated DNA and comparative analysis with a reference genome then facilitates the identification and quantification of methylation sites at the single-nucleotide level.
The principle of RRBS is predicated on two fundamental steps. Firstly, the genomic DNA is subjected to restriction endonuclease digestion. This enzymatic action fragments the DNA and preferentially retains regions abundant in CpG sites. These fragments are then subjected to terminal repair and ligation of sequencing adaptors to prepare them for downstream processing. Secondly, bisulfite conversion is performed, which forms the basis for distinguishing between methylated and unmethylated cytosines during sequencing and subsequent data analysis. The principle of RRBS is based on the enrichment of specific regions of the genome and bisulfite transformation, as follows:
Enrichment in specific regions: Genomic DNA is cut by restriction endonuclease (such as MspI), which recognizes CCGG sequence and cuts it, so that fragments rich in CpG islands are retained, because CpG islands often contain multiple CCGGs locus. This can reduce the complexity of genome and enrich the target region.
Terminal repair and linker connection: the cut DNA fragment is terminal repaired, sticky ends are added, and a specific sequencing linker is connected for subsequent PCR. amplificationAnd sequencing.
Sulfite transformation: When DNA is treated with sulfite, unmethylated cytosine (C) is transformed into uracil (U), while methylated cytosine remains unchanged.
PCR amplification and sequencing: The transformed DNA was used as a template for PCR amplification, in which U corresponds to thymine (T). After sequencing, the results were compared with the reference genome, and the methylation sites were analyzed.
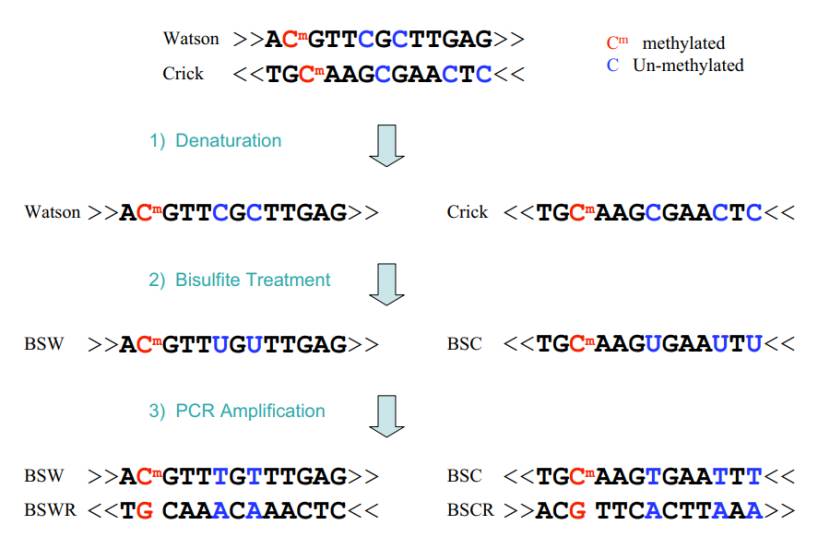 Principle of RRBS (Xi et al., 2009)
Principle of RRBS (Xi et al., 2009)
The RRBS process from sample processing to data interpretation helps the research of DNA methylation. First, genomic DNA was extracted from the sample, and after quality inspection, it was cut by restriction endonuclease to enrich CpG-rich regions. Repair the end of the cut fragment, add A tail and connect sequencing linker to construct the library. Subsequently, bisulfite treatment changed unmethylated cytosine into uracil, while methylation remained unchanged. Then PCR amplification, purification of the product and sequencing on the computer.
Sample preparation and initial processing
Genomic DNA extraction from the source material (cells or tissues) is the initial step. Stringent quality control measures, including agarose gel electrophoresis and ultraviolet spectrophotometry, are employed to ensure the integrity and purity of the DNA. Any sample exhibiting significant degradation or contamination is typically excluded from further analysis.
The extracted DNA is then digested with the chosen restriction enzyme. The digestion conditions are carefully optimized to ensure efficient cleavage at the target sites and maximal enrichment of CpG-rich fragments.
After digestion, the DNA fragments undergo terminal repair to generate blunt ends and addition of an adenine tail. This modification is essential for the subsequent ligation of sequencing adaptors.
Sequencing libraries are constructed by ligating the A-tailed DNA fragments with methylation-specific sequencing adaptors. Size selection is then performed to isolate fragments of the desired length range, which are further purified to remove any residual contaminants or unwanted by-products.
Bisulfite treatment and amplification
The purified DNA fragments are treated with bisulfite. This chemical treatment converts unmethylated cytosines to uracil, while methylated cytosines are resistant to this conversion. The reaction conditions are meticulously controlled to ensure complete and consistent conversion of unmethylated cytosines.
PCR amplification is carried out using the bisulfite-treated DNA as a template. The amplification primers are designed to anneal specifically to the converted and non-converted regions, allowing for selective amplification of the target DNA fragments. The PCR products are then purified to remove any residual primers, dNTPs, or enzymes.
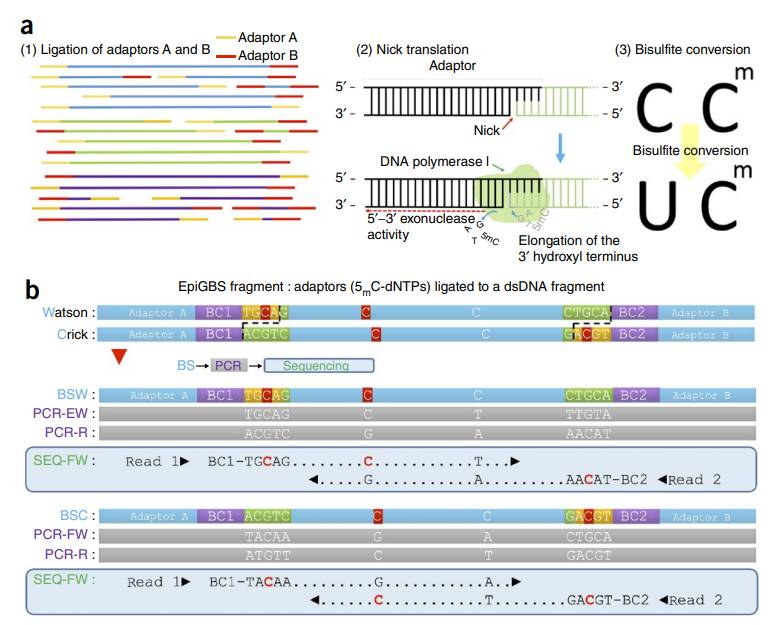 Method design of RRBS (Thomas et al., 2016)
Method design of RRBS (Thomas et al., 2016)
Bioinformatics analysis pipeline
Quality assessment of the sequencing data is performed using software tools such as FastQC. This initial analysis evaluates parameters such as base quality scores, sequence length distribution, and the presence of any adapter contamination or low-quality reads. Any sequences failing to meet the predefined quality thresholds are filtered out.
The processed sequences are aligned to the reference genome using alignment tools like Bismark or BSMAP. These tools are specifically designed to handle bisulfite-treated sequences and account for the C-to-T conversion of unmethylated cytosines.
Methylation levels at individual CpG sites are calculated based on the alignment results. The ratio of methylated CpG sites to the total number of CpG sites at a given locus is determined, providing a quantitative measure of methylation status.
Differential methylation analysis is conducted by comparing methylation levels between different samples or experimental groups. Software packages such as methylKit and edgeR are commonly utilized for this purpose. These tools employ statistical algorithms to identify differentially methylated regions (DMRs) or sites (DMPs) with a high degree of confidence.
Functional annotation of genes associated with the identified DMRs or DMPs is performed. This involves mapping the genomic locations of these regions to gene promoters, exons, introns, or intergenic regions. Enrichment analysis using databases such as GO and KEGG is then carried out to gain insights into the biological processes and signaling pathways potentially affected by the differential methylation events.
Visualization of the data is an integral part of the analysis pipeline. Using programming languages such as R and associated packages like ggplot2 and circlize, the data is presented in the form of heatmaps, boxplots, volcano plots, or Circos plots. These visual representations facilitate the identification of trends, patterns, and relationships within the data, enhancing the interpretability of the results.
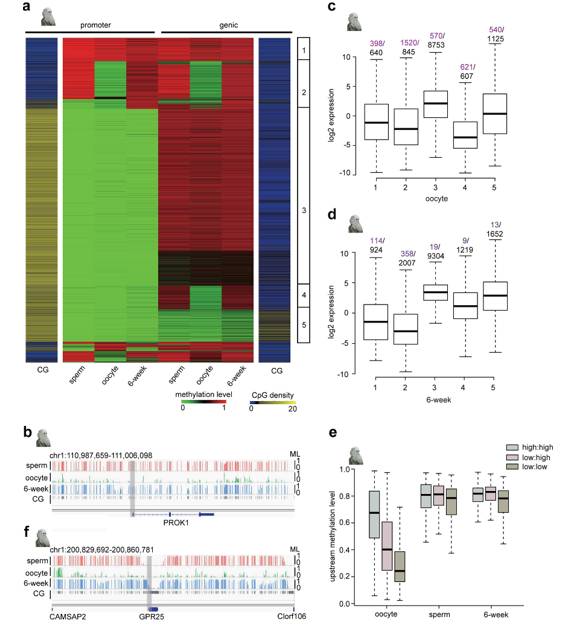 Oocyte-specific hypomethylated genic and intergenic regions (Li et al., 2018)
Oocyte-specific hypomethylated genic and intergenic regions (Li et al., 2018)
Service you may intersted in
If you want to learn more about the RRBS, please refer to:
RRBS has obvious advantages and is unique in the study of DNA methylation. In terms of cost, by sequencing specific CpG-rich regions, it can reduce the complexity of genome, reduce the amount of sequencing, greatly cut costs and achieve high cost performance. In terms of resolution, it can be accurate to the level of single nucleotide, clearly present methylation state, and analyze the key regulatory regions of genes in detail. In terms of sequencing depth, due to the small target area, high-depth sequencing can be realized and the data accuracy is high. The interpretation of biological meaning is simple, and the focused region is closely related to the regulation of gene expression, which makes the detection results more easily related to biological functions. In addition, the technology is mature, and the experimental operation and data analysis process are stable, which is conducive to wide application. Advantages of RRBS are as follows:
High cost-effectiveness: RRBS offers a cost-efficient alternative to whole-genome bisulfite sequencing (WGBS). By focusing on the CpG-rich regions, which typically account for only a small fraction (approximately 1%) of the genome, RRBS significantly reduces the sequencing volume and cost while maintaining a high level of data accuracy and resolution in the regions of interest.
High resolution: The ability to detect DNA methylation at the single-nucleotide level provides a detailed view of the methylation landscape within CpG islands, promoters, and other regulatory regions. This high-resolution analysis is crucial for understanding the fine-grained regulation of gene expression and identifying potential epigenetic biomarkers.
Mature experimental process: RRBS has a well-established experimental workflow. The combination of restriction enzyme digestion and bisulfite sequencing has been refined over time, and there are standardized procedures and guidelines available for each step of the process. This makes it accessible to researchers with varying levels of expertise and facilitates reproducibility across different laboratories.
The biological significance is clear: The regions targeted by RRBS are highly relevant to gene regulation. Changes in methylation status within these regions are often associated with significant alterations in gene expression and cellular function. Therefore, the results obtained from RRBS studies can be directly linked to biological processes and disease mechanisms, providing valuable insights into the underlying biology.
Although RRBS is valuable, it has obvious disadvantages. Its coverage area is limited, only for specific areas rich in CpG, leaving out methylation information of other parts of the genome, making it difficult to see the whole picture. Depending on the restriction site, some areas without suitable sites can not be detected, resulting in information loss. The initial amount of samples is high, which requires microgram DNA, and the application of rare or difficult-to-obtain samples, such as micro clinical biopsy and paleontological samples, is limited. In terms of data, due to the complexity of non-genome sequencing, splicing and analysis, it is difficult to be as intuitive and comprehensive as genome-wide data, and it is difficult to adapt when it is integrated with other genome-wide research. disadvantages of RRBS are as follows:
Limited coverage area: One of the primary limitations of RRBS is its inability to provide a comprehensive view of the entire genome's methylation status. The reliance on restriction enzyme recognition sites restricts the analysis to regions containing these specific sequences, potentially missing important methylation changes in other parts of the genome.
Restriction of restriction sites: The success of RRBS is highly dependent on the presence and distribution of appropriate restriction sites within the genome. If certain regions lack the target recognition sequences, they will not be enriched or sequenced, leading to incomplete or biased data.
High initial sample size: RRBS typically requires a relatively large amount of starting genomic DNA, usually in the microgram range. This can pose a challenge when working with limited or precious samples, such as those obtained from small clinical biopsies or rare cell populations.
There is a certain degree of DNA degradation: Bisulfite treatment can cause some degree of DNA degradation, especially in longer DNA fragments. This can affect the quality and coverage of the sequencing data, potentially reducing the ability to detect methylation events in larger genomic regions.
RRBS data analysis aims to reveal DNA methylation patterns and functions. The original sequencing data were first evaluated for quality, and the low-quality and polluted sequences were removed. Subsequently, that proces sequence is aligned with a reference genome by a specific tool to determine its position. Based on the comparison results, the methylation status of CpG sites was counted and the level was calculated.
In order to explore the biological significance, different samples will be compared and different methylation regions and sites will be identified. Then, the function of related genes is annotated, the distribution in genomic elements is clarified, and the biological processes and signal pathways involved are revealed by enrichment analysis. The whole analysis process is closely linked, which provides key insights for understanding the role of methylation in biological processes. RRBS data analysis is used to explore DNA methylation patterns, and the main steps are as follows:
Data preprocessing: Use FastQC to evaluate the quality of original sequencing data and remove low-quality bases, linkers and contaminated sequences. Tools such as Trim Galore can filter and trim data and improve data quality.
Sequence comparison: Compare that processed sequence with the reference genome to determine its position in the genome. Bismark, BSMAP and other tools can compare the sequence after bisulfite treatment to the reference genome, considering the C-to-T transformation.
Methylation level calculation: According to the comparison results, the methylation status of each CpG site is counted and the methylation level is calculated, that is, the number of methylated C is divided by the total number of C in this site.
Differential methylation analysis: Compare methylation levels of different samples and identify differential methylation regions (DMR) or sites (DMP). MethylKit, edgeR and other tools can identify DMR and DMP, and analyze the significant difference.
Functional annotation and enrichment analysis: Annotate the functions of genes related to differential methylation and determine their distribution in genomic elements, such as promoters and genomes. Use GO,KEGGThe biological process and signal pathway of differential methylation were revealed by database enrichment analysis.
Visualization analysis: Draw heat map, box map, volcano map and Circos map with r language ggplot2, circlize and other packages, visually display methylation data, and mine characteristics and laws.
RRBS is widely used. In disease research, methylation biomarkers of cancer and other diseases can be tapped to assist diagnosis, and the pathogenesis can be analyzed, providing key clues for the study of nervous system diseases. In the field of developmental biology, it can reveal the mystery of methylation regulation of cell differentiation and tissue formation in embryonic development. In drug research and development, RRBS helps to clarify the methylation status of drug targets and promote targeted drug development. In botanical research, by analyzing the methylation patterns of plants in response to environmental changes and different development stages, it provides a basis for crop stress resistance improvement and variety optimization.
Disease diagnosis and research: In cancer research, RRBS has been instrumental in identifying methylation changes in tumor suppressor genes and other cancer-related genes. These methylation alterations can serve as potential biomarkers for early cancer detection and diagnosis. In neurological disorders such as Alzheimer's disease, RRBS has been used to explore the role of DNA methylation in disease pathogenesis by comparing methylation profiles between patients and healthy controls.
Developmental biology: RRBS has provided valuable insights into the epigenetic regulation of embryonic development. By analyzing the methylation dynamics during cell differentiation and tissue formation, researchers can better understand the molecular mechanisms underlying these processes and how they are controlled by DNA methylation.
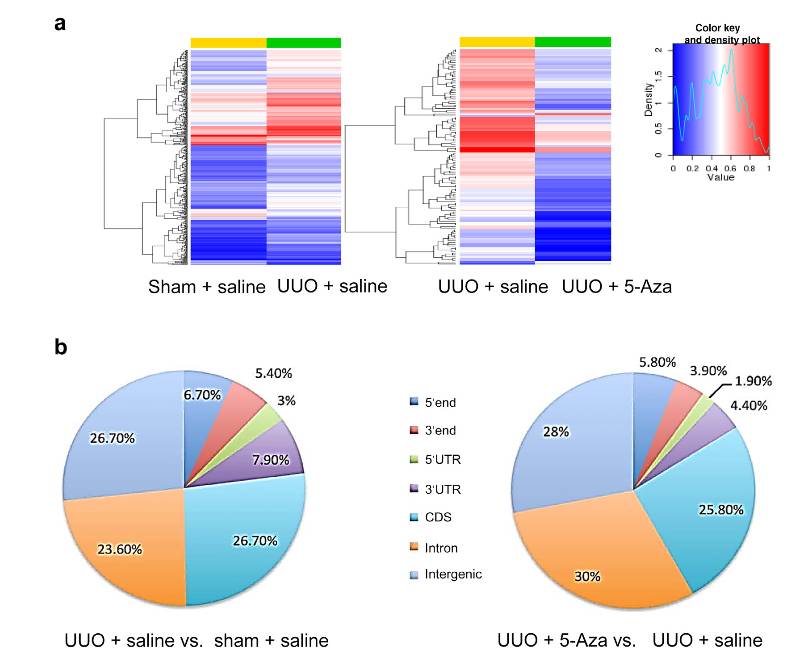 Profiling of DNA methylation in unilateral ureter obstruction (UUO) kidneys (Xiao et al., 2024)
Profiling of DNA methylation in unilateral ureter obstruction (UUO) kidneys (Xiao et al., 2024)
Drug research and development: In the field of drug discovery, RRBS helps in characterizing the methylation status of drug targets. This information can guide the development of targeted therapies, such as methyltransferase inhibitors, by identifying genes with abnormal methylation patterns that could be potential therapeutic targets.
Botanical research: In plant biology, RRBS has been applied to study the epigenetic responses of plants to environmental stresses and developmental cues. By analyzing DNA methylation changes in different plant tissues and under various conditions, researchers can identify genes involved in stress tolerance and plant development, which can be exploited for crop improvement.
In conclusion, RRBS represents a powerful tool in the epigenetic research toolkit, with unique advantages and limitations. Its applications span multiple disciplines, from basic biological research to translational medicine and agriculture. As the field of epigenetics continues to evolve, further refinements and improvements in RRBS technology are expected to enhance its utility and expand its potential applications. Future research directions may include the development of novel restriction enzymes or alternative enrichment strategies to overcome the current limitations and improve the comprehensiveness of the analysis. Additionally, integrating RRBS data with other omics data sources will provide a more holistic understanding of the complex regulatory networks underlying biological processes and disease states.
If you want to learn more about the RRBS, please refer to:
References
Terms & Conditions Privacy Policy Copyright © CD Genomics. All rights reserved.