




We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
DNA methylation is an important change in the genome that controls gene activity without changing the DNA sequence. This change involves adding a methyl group (CH₃) to the DNA, usually at certain points in the DNA sequence. DNA methyltransferases (DNMTs) catalyze this addition by transferring a methyl group from S-adenosylmethionine (SAM) to cytosine. As a fundamental regulator of gene activity, DNA methylation is integral to numerous cellular processes, including differentiation, development, and the pathogenesis of diseases.
In mammals, including humans, DNA methylation typically manifests as 5-methylcytosine (5-mC), which functions to repress gene expression either by obstructing transcription factor binding or by promoting the formation of a condensed chromatin structure. Although methylation can also occur on other bases such as adenine (N6-methyladenine) and guanine (7-methylguanine), these occurrences are less common and not as extensively characterized as cytosine methylation.
The importance of DNA methylation extends beyond mere biological functions; it is a stable and potentially heritable modification that modulates phenotypic trait expression. Consequently, it is a cornerstone of epigenetic science. The dynamic regulation of DNA methylation patterns holds significant consequences for both health and disease, particularly in the contexts of cancer, neurodegenerative disorders, and developmental diseases.
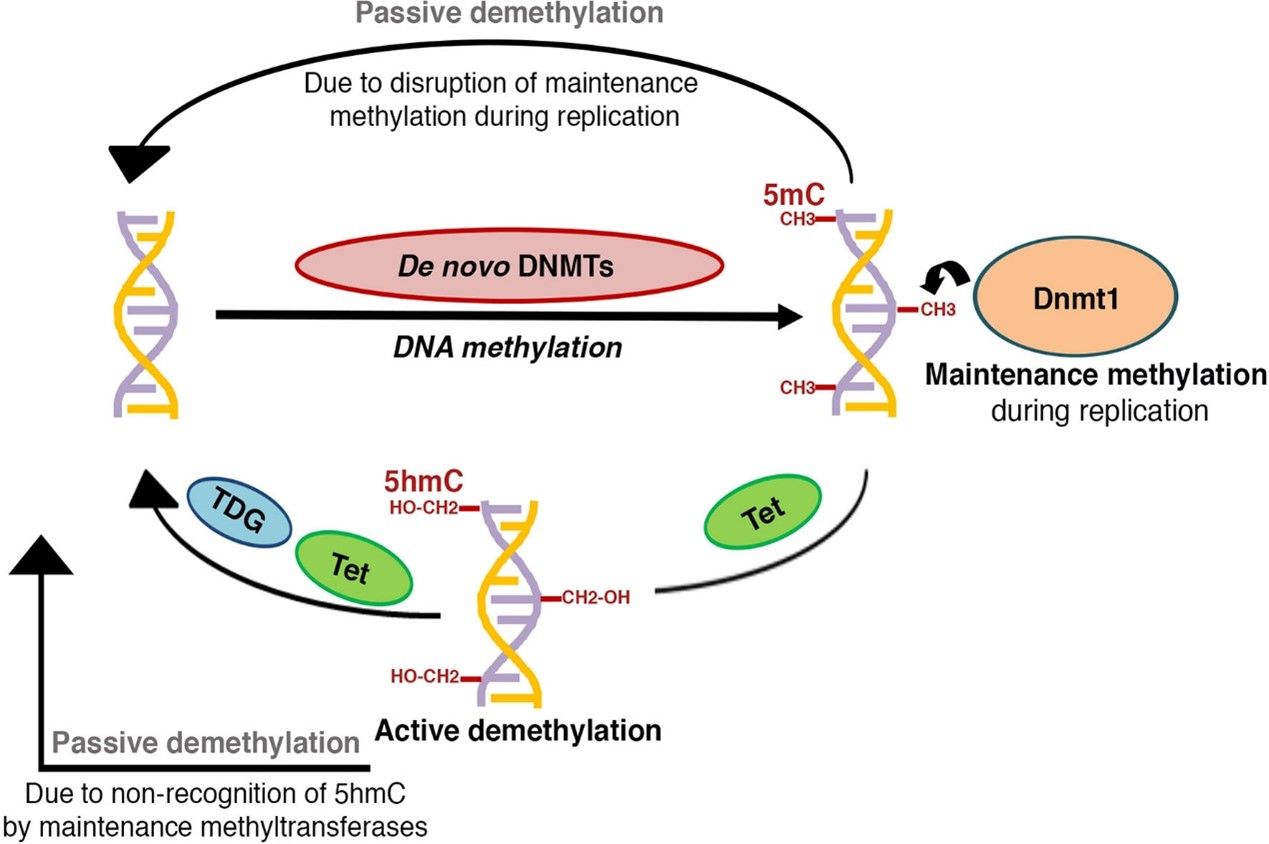 Diagram of DNA methylation. (Parveen, et al., Frontiers in Endocrinology, 2021)
Diagram of DNA methylation. (Parveen, et al., Frontiers in Endocrinology, 2021)
DNA methylation is a pivotal modification within the epigenome, primarily occurring at CpG sites. It involves the covalent attachment of a methyl group to the 5th carbon atom of the cytosine ring. This essential epigenetic modification is orchestrated by the DNA methyltransferase enzyme family, comprising DNMT1, DNMT3A, and DNMT3B, which specifically target CpG dinucleotides to methylate the cytosine base.
DNA methylation is guided by two principal mechanisms:
1. De novo Methylation: This process is predominantly active during the early stages of development, particularly within embryonic cells, where new methylation patterns are established. The enzymes DNMT3A and DNMT3B are critical in adding methyl groups to previously unmethylated DNA, a fundamental initial step in determining cellular identity during differentiation.
2. Maintenance Methylation: During DNA replication, the newly synthesized DNA strand is initially unmethylated. DNMT1 detects hemimethylated DNA, characterized by methylation marks present only on the original strand, and subsequently methylates the complementary strand. This mechanism ensures the continuity of methylation patterns across successive cell divisions, playing a crucial role in maintaining both the fidelity of epigenetic marks and the integrity of the epigenome, thereby preserving cell-specific gene expression profiles.
The end result of methylation is the creation of 5-methylcytosine (5-mC), which helps turn off certain genes. It obstructs the binding of transcriptional activators to gene promoter regions, thereby inhibiting gene expression. In addition, methylated DNA functions as a binding platform for methyl-binding proteins (MBPs), which foster the establishment of a repressive chromatin environment, further suppressing gene transcription.
This section highlights the crucial role of DNA methylation in gene regulation, disease development, and epigenetic inheritance, emphasizing its importance in development, disease, and intergenerational transmission.
DNA methylation is pivotal for gene expression regulation during development. During embryogenesis, a distinct methylation landscape is established, influencing cell fate decisions. For instance, in stem cells, the equilibrium between methylation and demethylation dictates pluripotency retention or differentiation into specialized cell lineages. This meticulous regulation ensures the fidelity of gene expression programs pertinent to each cell type.
Furthermore, DNA methylation underlies X-chromosome inactivation in females. In mammals, one X chromosome is randomly silenced during development through extensive methylation, ensuring monoallelic expression of X-linked genes.
Aberrant methylation patterns are implicated in diseases, particularly cancer. Hypermethylation of tumor suppressor genes can silence these critical regulators, facilitating oncogenic transformation, while hypomethylation of oncogenes may result in unwarranted activation. Such dysregulation of methylation is evident in various cancers, including breast, lung, and colorectal neoplasms.
Methylation changes also play roles in neurodegenerative disorders like Alzheimer's disease, where altered methylation may disrupt essential genes for synaptic functionality and memory. Additionally, aberrant methylation patterns are associated with autoimmune conditions, cardiovascular diseases, and psychiatric disorders, underscoring their significance in cellular equilibrium and functionality.
A compelling attribute of DNA methylation is its potential transmissibility across generations as an epigenetic mark. Unlike permanent genetic mutations, epigenetic modifications, such as methylation, may be inherited without changes to the DNA sequence itself. Observations in both plants and animals suggest that epigenetic inheritance influences inheritance and evolutionary patterns. External environmental factors, including nutrition, stress, and toxic exposures, can modify methylation landscapes, thereby impacting phenotypic expression without genomic alteration.
DNA acetylation is a pivotal post-translational modification, extensively implicated in the regulation of gene expression. It involves the covalent attachment of an acetyl group (CH₃CO-) to histone proteins associated with DNA, instigating alterations in chromatin architecture and modulating gene activity. Acetylation is predominantly recognized as an activating modification, renowned for its potency and substantial impact on gene expression. This modification typically promotes gene expression by acetylating lysine residues in histone tails within accessible chromatin regions, a process often initiated by specific transcriptional activators, including certain nutritional factors, interleukin-4, and heparin-binding epidermal growth factor.
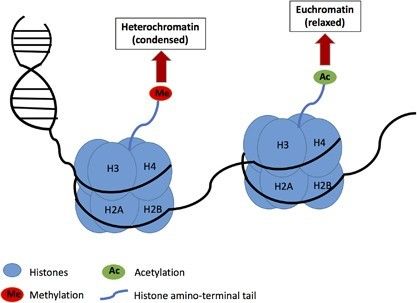 Schematic drawing of histone methylation and acetylation in relation to chromatin remodeling. (Kim, S., et al., Exp Mol Med, 2017)
Schematic drawing of histone methylation and acetylation in relation to chromatin remodeling. (Kim, S., et al., Exp Mol Med, 2017)
DNA methylation and acetylation represent reversible epigenetic modifications, enabling cellular responsiveness to dynamic growth and developmental signals. While both modifications regulate gene expression by adding chemical groups, their mechanistic pathways markedly diverge. DNA methylation involves the addition of methyl groups to cytosine bases, leading to a condensed chromatin state that hinders transcription factor binding, thereby reducing gene transcriptional activity. In contrast, acetylation of histone tails results in a relaxed chromatin configuration that facilitates gene activation and expression, thus playing a crucial role in the regulatory dynamics of genes.
Moreover, an elaborate interplay between DNA methylation and acetylation exists, characterized by reciprocal influences. Although the sites and mechanisms of these modifications are not completely overlapping, they can mutually affect each other's attachment and distribution. Current research indicates that these two epigenetic mechanisms often act synergistically, collaboratively orchestrating gene expression and performing essential roles within the intricate architecture of biological systems.
The field of DNA methylation analysis is experiencing rapid progress, with a variety of methodologies emerging to detect and quantify this vital epigenetic modification. Broadly, these methodologies are classified into those that evaluate global methylation levels and those that target specific methylation sites.
HPLC remains a prevalent technique for estimating global DNA methylation levels. In this method, DNA is hydrolyzed into nucleosides, and 5-methylcytosine (5-mC) is distinguished from other nucleosides through reverse-phase chromatography. This facilitates the quantification of 5-mC content genome-wide. An enhanced variant, HPLC coupled with mass spectrometry (HPLC-MS), offers increased sensitivity and specificity in methylation detection.
MSREs specifically recognize and cleave unmethylated cytosine residues, whereas methylated cytosines remain unaltered. This characteristic is harnessed to detect methylation at particular genomic loci. Post-digestion, PCR amplification or electrophoresis determines the methylation status of DNA fragments. Nevertheless, this technique is constrained by the availability of appropriate restriction enzymes.
Bisulfite treatment is a key method for detecting DNA methylation. It keeps methylated cytosines intact and changes unmethylated ones. Then, the differences are detected using PCR or sequencing to study specific sites. Bisulfite sequencing is renowned for its precision, offering single-base resolution of DNA methylation patterns.
MeDIP employs antibodies that selectively bind to methylated DNA, followed by sequencing to chart methylation patterns across the genome. This approach is particularly effective for large-scale, genome-wide methylation studies and can be integrated with next-generation sequencing to achieve high-resolution mapping.
Innovative technologies such as Single-Molecule Real-Time (SMRT) sequencing and Oxford Nanopore sequencing enable the real-time, direct detection of DNA methylation. These methods provide long-read sequencing with high single-base resolution, facilitating the evaluation of methylation across entire genomes without necessitating bisulfite treatment. However, these techniques are currently under development, often involving higher costs and lower throughput relative to conventional sequencing methods.
Collectively, these cutting-edge techniques furnish robust instruments for the investigation of DNA methylation, thus advancing our understanding of epigenetic modifications across developmental pathways, disease states, and personalized medicine paradigms.
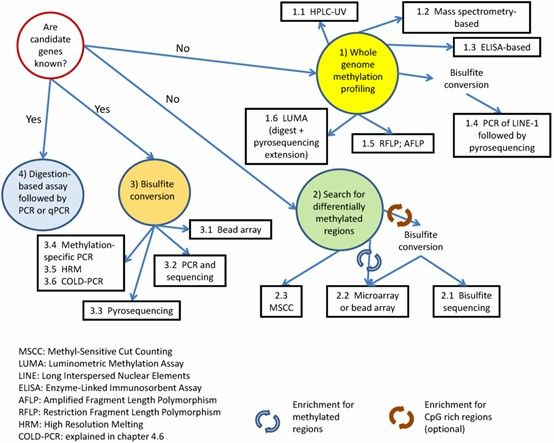 Algorithm for choosing a suitable method for DNA methylation analysis. (Kurdyukov, et al., Biology, 2016)
Algorithm for choosing a suitable method for DNA methylation analysis. (Kurdyukov, et al., Biology, 2016)
CD Genomics offers a comprehensive suite of advanced DNA methylation sequencing services, enabling precise analysis of epigenetic modifications for a wide range of applications:
DNA methylation research is revolutionizing medical practices, providing valuable insights into cancer diagnosis, personalized medicine, and the development of epigenetic therapies.
DNA methylation patterns serve as potent biomarkers for cancer detection, diagnosis, and prognosis. Specific epigenetic methylation signatures, detectable through DNA methylation sequencing, correlate with distinct cancer types and can be discerned in body fluids such as blood, saliva, or urine. For instance, MLH1 gene promoter hypermethylation is a colorectal cancer diagnostic marker, while BRCA1 promoter hypermethylation is associated with breast cancer. The non-invasive detection of these methylation changes heralds improvements in early cancer detection and patient prognoses.
Deciphering individual methylation profiles opens pathways for personalized medical interventions. DNA methylation patterns govern patient responses to treatments like chemotherapy or immunotherapy. Certain cancers exhibiting distinct methylation profiles may show heightened susceptibility to specific therapeutic agents. Tailoring interventions based on these epigenetic markers enhances treatment efficacy and minimizes adverse effects.
The burgeoning field of epigenetic therapies targets the epigenome for therapeutic benefit. DNA methyltransferase inhibitors (DNMTi), such as 5-azacytidine, are already employed to combat certain cancers by reversing aberrant methylation patterns. These therapies endeavor to restore normative gene expression, thereby reactivating tumor suppressor genes and reinstating their protective roles against malignancies.
The field of DNA methylation research is on the brink of transformation, holding significant promise for therapeutic innovation and medical advancements. As cutting-edge methodologies, including advanced DNA methylation sequencing, continue to develop, our comprehension of its integral roles in development, disease pathology, and the maintenance of the epigenome is poised to expand dramatically. There is an increasing focus within the research community on identifying specific DNA methylation biomarkers that facilitate earlier and more precise diagnoses across a variety of diseases.
Combining DNA methylation data with other genetic information will help us better understand gene regulation. This systems-level perspective may catalyze the development of targeted epigenetic therapies specifically designed to modify distinct DNA methylation patterns. Such therapeutic strategies have the potential to address a diverse array of conditions, including various cancers, autoimmune disorders, and neurodegenerative diseases, by rectifying dysregulated epigenetic mechanisms.
With rapid advancements in the methodologies surrounding DNA methylation studies, the medical field is poised for revolutionary changes. Organizations such as CD Genomics are leading these innovations, laying the groundwork for breakthroughs that could fundamentally alter therapeutic practices. By leveraging the potential of epigenetic modifications, researchers are on the cusp of transforming the diagnosis, treatment, and prevention of myriad diseases, with broad implications for enhancing patient outcomes and public health at large.
References
Terms & Conditions Privacy Policy Copyright © CD Genomics. All rights reserved.