





We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
The prevailing paradigm of the central law posits that RNA functions exclusively as a conduit between DNA and proteins, transmitting information. However, the advent of epitranscriptomics has precipitated a paradigm shift in this understanding. The presence of over 170 distinct chemical modifications on the RNA molecule has unveiled a complex regulatory system that functions as a "molecular switch," thereby dictating the spatial and temporal specificity of gene expression. These modifications, ranging from tRNA stability to early cancer diagnosis, are redefining the boundaries of our understanding of life regulation. The integration of multi-omics analysis and AI-driven epigenetic annotation tools, enabled by breakthroughs in long-read sequencing and single-cell epigenomics, is propelling this field to the forefront of precision medicine.
Following a decade of m6A's preeminence in the epitranscriptomic domain, researchers have come to recognize the extensive diversity of RNA modifications. This encompasses a range of modifications, from m7G, which regulates translational precision, to pseudouridine (Ψ), which enhances RNA stability, and including m5C and m1A, the episodic sentinels of mitochondrial RNA. These modifications collectively reshape patterns of gene expression through dynamic chemical labeling. This review will synthesize the most recent advancements in m7G RNA methylation sequencing, PA-Ψ-seq, and other state-of-the-art technologies to elucidate the role of these "dark substances" as crucial regulators of disease.
m7G: "molecular proofreaders" guarding the quality of translations
In the field of RNA modifications, m7G (7-methylguanosine) has been recognized as a "structural stabilizer" of tRNAs and rRNAs. Employing m7G RNA methylation sequencing technology, researchers have determined that m7G modifications, which are extensively present on the anticodon loops of mammalian tRNAs, can impede cleavage by nucleic acid exonucleases and thereby maintain the accuracy of the translation process. In the context of acute myeloid leukemia, a mutation in the METTL1 enzyme has been observed to result in a decline in the level of the tRNA m7G modification. This, in turn, has been shown to trigger ribosomal shifting errors and to lead to a substantial accumulation of aberrant truncations of the oncogenic protein MYC. These findings suggest that the methylation status of tRNA modification enzyme genes could be monitored using targeted DNA methylation analysis techniques, such as EpiTYPER, to potentially develop novel prognostic markers for hematologic tumors.
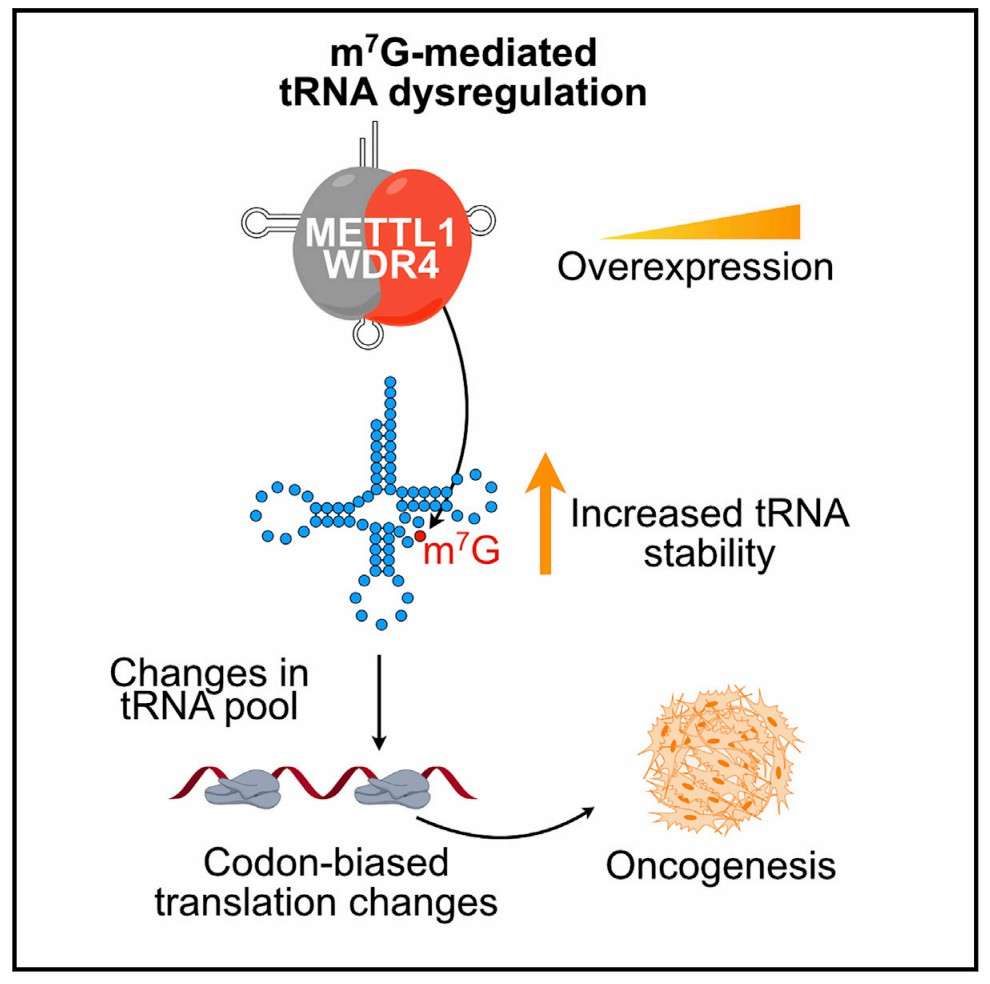 METTL1-mediated m7 G modification of Arg-TCT tRNA drives oncogenic transformation (Esteban et al., 2021)
METTL1-mediated m7 G modification of Arg-TCT tRNA drives oncogenic transformation (Esteban et al., 2021)
Pseudouridine: a "buffer" for cellular stress
Pseudouridine (Ψ) is one of the most prevalent modifications in RNA, with its distinctive C-C glycosidic bond enhancing the thermal stability of RNA molecules. Utilizing a transcriptome-wide scan of rRNAs employing PA-Ψ-seq technology, scientists have identified that the level of nucleolar small RNA (snoRNA)-directed pseudouridylation is markedly augmented under hypoxic conditions. For instance, in hepatocellular carcinoma cells, the study found that the level of SNORD78-mediated Ψ modification at position 4321 of the 28S rRNA maintains the translational efficiency of ribosomes in a hypoxic microenvironment and promotes the synthesis of angiogenic factors such as VEGF. This mechanism provides a theoretical basis for the development of RNA modification-targeted inhibitors. For instance, blocking the binding of specific snoRNAs to rRNAs by chemical probes (e.g., DM-seq) may inhibit adaptive tumor survival.
m5C and m1A: "episodic sentinels" of mitochondrial RNAs
Research on mitochondrial RNA modifications had been hindered by technical limitations; however, breakthroughs in m5C RNA-seq and m1A RNA methylation analysis have transformed the field. In an Alzheimer's disease model, researchers discovered that the deletion of m5C modification of neuronal mitochondrial tRNA results in impaired assembly of respiratory chain complex I, triggering a collapse of energy metabolism. Furthermore, the application of acRIP-seq technology has revealed an aberrant accumulation of m1A modifications in the mitochondrial 16S rRNA of brain tissues from individuals diagnosed with Parkinson's disease. This accumulation hinders the binding of ribosomes to mRNA, thereby accelerating the degeneration of dopaminergic neurons. These findings suggest that the combined application of DNA hydroxymethylation analysis (e.g., 5hmC-Seal Seq) and RNA modification detection may provide a two-dimensional marker for the early diagnosis of neurodegenerative diseases.
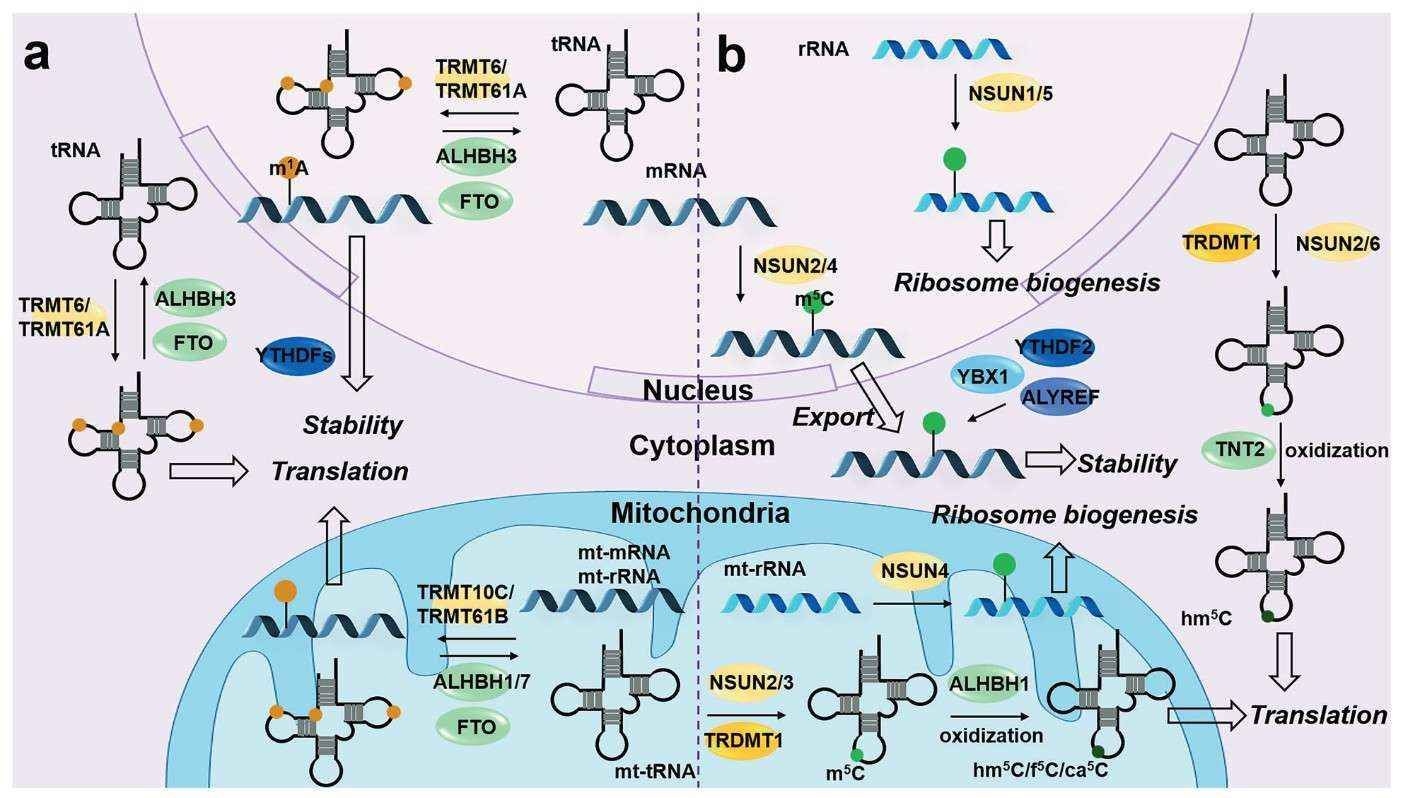 Dynamic m1A and m5C modification on RNAs (Wang et al., 2023)
Dynamic m1A and m5C modification on RNAs (Wang et al., 2023)
Service you may intersted in
Learn More:
A comprehensive analysis of the spatiotemporal dynamics of RNA modifications necessitates the advancement of conventional technologies to enhance their resolution and sensitivity. Nanopore direct sequencing facilitates the detection of modifications at the single-molecule level, while chemical labeling techniques (e.g., DM-seq) enable the identification of rare modifications at an abundance of one in ten thousand. Integrative multi-omics analyses transform discrete data into meaningful biological insights. This section will demonstrate how these technologies are driving the transition from basic research to precision medicine through clinical study examples.
Nanopore direct sequencing: rewriting the "rules of the game" for modification detection
Conventional approaches to RNA modification detection entail the use of antibody enrichment or chemical processing. In contrast, nanopore direct RNA sequencing (ONT Direct RNA Sequencing) represents a revolutionary advance in the field. This method facilitates the direct reading of full-length RNA sequences without the need for fragmentation, thereby enabling the direct identification of modification sites, including m6A, m5C, and others, through the analysis of current signaling differences. In the domain of neuroscience, this technology unveils the dynamics of m6A modification of tau protein mRNAs: when neuronal synaptic activity is enhanced, FTO enzymes are recruited to erase the m6A tag locally at the synapse, thereby promoting local translation of tau proteins, a process that is closely related to memory consolidation. Integration of chromatin accessibility data (ATAC-seq) by the researchers further revealed that synaptic activity alters the chromatin opening state in a simultaneous manner and activates the expression of m6A methyltransferases (e.g., METTL3), thereby creating a positive feedback loop of epistatic regulation.
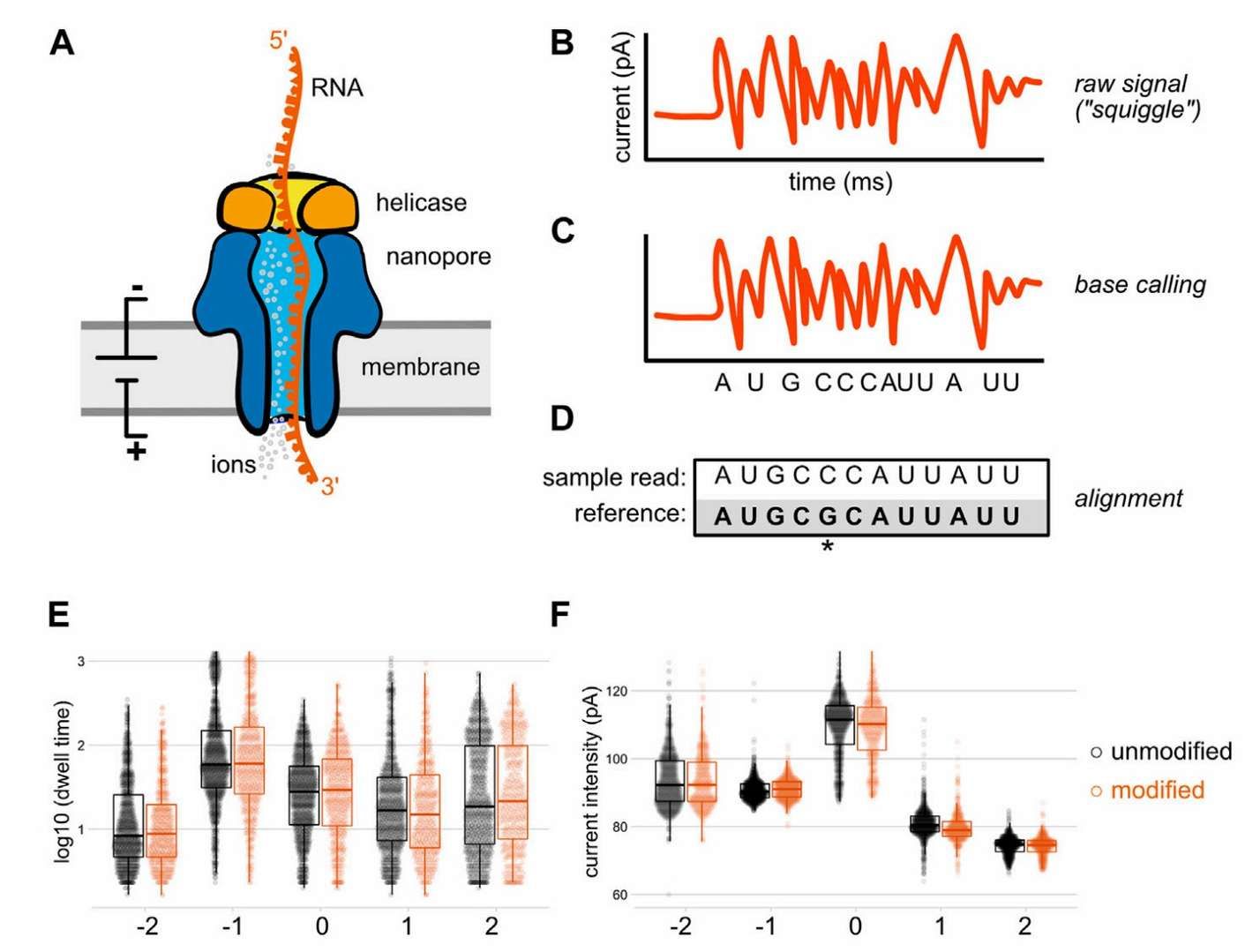 Direct RNA sequencing by nanopore (White et al., 2022)
Direct RNA sequencing by nanopore (White et al., 2022)
Chemical labeling techniques: capturing low abundance modified "dark matter"
In the context of rare modifications, such as m1A, 2'-O-methylation, with a frequency of less than 1 in 10,000, traditional antibody-based methods, including MeRIP-Seq, frequently prove ineffective. Conversely, DM-seq technology has been shown to enhance detection sensitivity by two orders of magnitude. This enhancement is achieved through the specific insertion of an acrylamide probe into the m1A site, in conjunction with click chemistry, which introduces biotin labeling. In the context of lung cancer research, this technology has facilitated the discovery that tumor cells exhibit a selective translation of mRNAs encoding oncogenes (e.g., KRAS) by augmenting the activity of the TRMT6/61A complex and elevating the level of m1A modification of tRNAs. This mechanism, termed "modification-preferred translation," unveils novel pathways for the development of targeted therapies based on epitomes. This mechanism of "modification-preferred translation" unveils a novel avenue for the development of epitranscriptome-targeted therapies.
Data integration: from massive information to biological insights
Single-technology assays are capable of revealing only a fraction of the total RNA modifications present; however, integrated multi-omics analysis is essential for comprehending their functions. For instance, the Epigenomic Peak Calling algorithm enables researchers to automatically differentiate authentic m6A sites from non-specific signals derived from MeRIP-seq data. Moreover, in conjunction with single-cell ATAC-seq (scATAC-seq) data, the chromatin localization of modifying enzymes can be traced during the process of cell differentiation. In the context of embryonic stem cell studies, this integration strategy has been instrumental in elucidating the role of METTL3 in maintaining the undifferentiated state of stem cells by recognizing open chromatin regions and specifically methylating mRNAs associated with pluripotency (e.g., NANOG).
Recent advancements in the field of RNA modification have led to significant progress in clinical translation, with potential applications in the treatment of metabolic diseases, early cancer screening, and the design of combination therapies across epigenetic levels. These developments have been made possible by the emergence of micro-sample technologies, such as EM-seq, and high-throughput assay platforms, including the Human Methylome Panel. This section aims to explore the potential of redefining the underlying logic of disease intervention by targeting RNA modifications.
FTO inhibitors: the "epigenetic brake" on obesity treatment
The overactivity of the m6A eraser, FTO, has been linked to energy metabolism imbalance. In animal experiments, the administration of small molecule inhibitors (e.g., FB23-2) has been observed to increase the m6A-modified level of leptin mRNA in adipocytes, prolong its half-life, and promote leptin protein secretion. Furthermore, the administration of FTO inhibitors to animal models of metabolic disease has been shown to elicit a "killing two birds with one stone" effect, concomitantly activating demethylation of the UCP1 gene and promoting the conversion of white to brown fat. This finding underscores the potential of FTO inhibitors as a promising therapeutic avenue for the treatment of metabolic diseases.
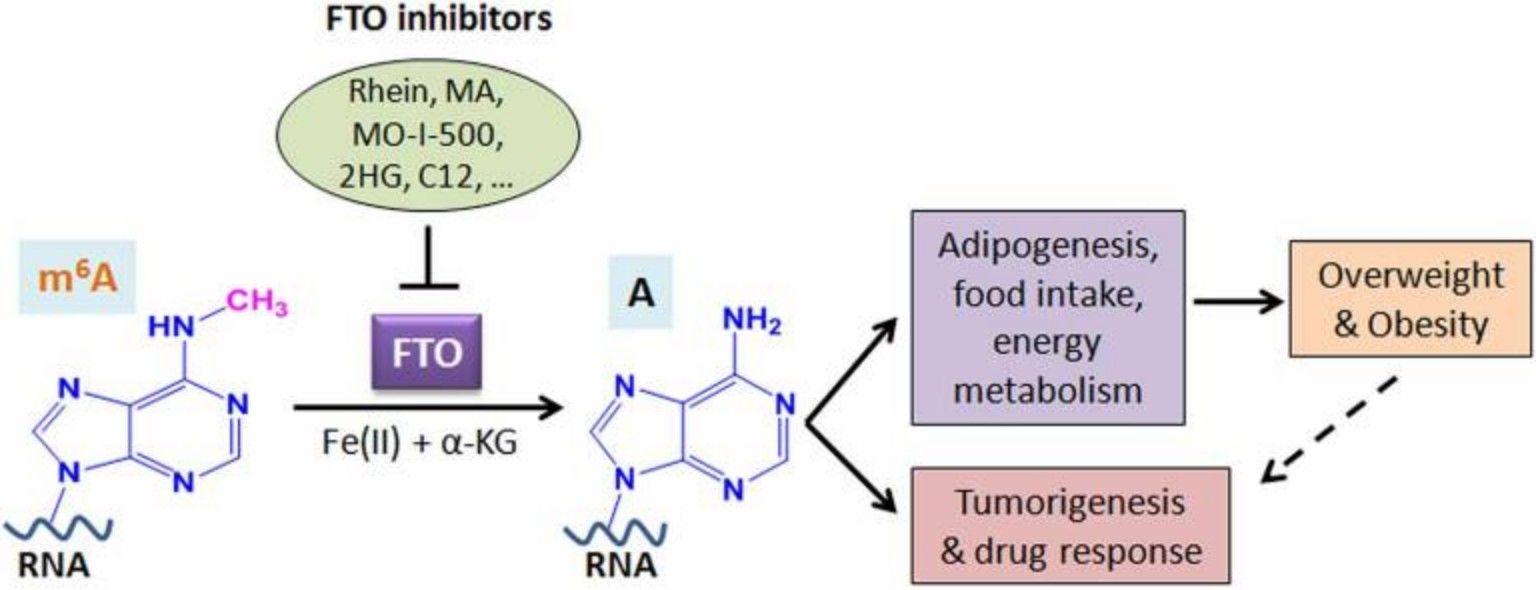 Schematic illustration of the roles of FTO in RNA m6A modification, overweight/obesity, and tumorigenesis/drug response (Deng et al., 2018)
Schematic illustration of the roles of FTO in RNA m6A modification, overweight/obesity, and tumorigenesis/drug response (Deng et al., 2018)
Liquid Biopsy: "Molecular Radar" for Early Cancer Screening
Methylation testing of circulating tumor DNA (ctDNA) in the blood is already a clinical practice, and analysis of exosomal RNA modifications may offer even earlier warning signals. Investigators have found that 5hmC modifications characteristic of plasma exosomal RNAs (e.g., 5hmC deletion of ALB mRNA) in patients with hepatocellular carcinoma can appear up to six months prior to the detection of tumors on imaging. When combined with the multidimensional analysis of ctDNA by the Human DNA Methylation Microarray, the sensitivity of early diagnosis of liver cancer can be increased from 68% to 89%. In addition, EM-seq technology enables the construction of genome-wide methylation profiles for trace samples, such as puncture biopsies, by optimizing the sulfite conversion process, which supports personalized treatment decision-making.
Cross-border convergence: a picture of the future of epimedicine
Cross-regulation of RNA modifications with other epitopes provides novel ideas for drug development. For example, in breast cancer treatment, the combination of DNA methyltransferase inhibitors (e.g., 5-aza) and m6A methylation inhibitors (e.g., STM2457) can synergistically activate the expression of oncogenes (e.g., BRCA1) and reverse chemoresistance in tumor cells. Furthermore, by leveraging the dynamics of histone modifications (e.g., H3K27me3) through Chromatin Immunoprecipitation Sequencing (ChIP-Seq), researchers have identified that RNA modifying enzymes can recruit Polycomb complexes to specific gene sites, thereby establishing a gene silencing network across epitopes. This cascade regulatory mechanism provides a theoretical foundation for the development of multi-target epigenetic drugs.
The field of epitranscriptomics is challenging the conventional boundaries of traditional molecular biology in several ways. For example, O8G-Seq has revealed oxidative stress markers, and Hi-C technology has resolved spatial associations between 3D genomes and RNA modifications. In the future, with the popularization of single-cell multi-omics technologies (e.g., scATAC-Seq combined with scRNA-seq) and the deep mining of massive data by AI-driven epigenetic annotation tools, we will have a clearer picture of the panorama of RNA modifications in development, aging, and disease.
References
Terms & Conditions Privacy Policy Copyright © CD Genomics. All rights reserved.