MeRIP-seq for Detecting RNA methylation: An Overview
Epitranscriptome pertains to the phenomenon wherein gene expression is modulated by chemical alterations on RNA without altering the RNA sequence. There exist over 100 types of intracellular RNA modifications, with the majority being conspicuous modifications on tRNA and other non-coding RNA, and only a few modifications on mRNA. The most abundant modification in mRNA is N6-methyladenosine (m6A), which partakes in all facets of the mRNA life cycle. Research has demonstrated that the primary function of m6A is to govern the stability of mRNA.
What is MeRIP-seq
Presently, high-throughput sequencing, colorimetry, and liquid chromatography-mass spectrometry (LC-MS) are the technical modalities for detecting m6A. Colorimetry and LC-MS can measure the overall m6A level of mRNA. LC-MS/MS employs tandem mass spectrometry based on liquid mass spectrometry to acquire molecular ion peaks and fragment ion peaks, and concurrently conducts qualitative and quantitative analyses of bases. However, these two methods are incapable of localizing m6A and are typically employed in the initial stages of research. Currently, methylated RNA immunoprecipitation sequencing (MeRIP-seq) is predominantly utilized in high-throughput sequencing and is one of the most prevalently used techniques for investigating m6A modification.
MeRIP-seq amalgamates methylated DNA immunoprecipitation (Medip) technology, RNA immunoprecipitation (RIP) technology, and RNA sequencing (RNA-seq) technology to precisely detect RNA methylation across the entire genome (or whole transcription group). The principle involves the specific recognition of m6A-modified antibodies to immunoprecipitate m6A-modified RNA fragments within cells. Through high-throughput sequencing analysis of the precipitated RNA fragments in conjunction with bioinformatics analysis, the modification of m6A in the entire genome can be systematically examined.
Principle of MeRIP-seq
Immunocoprecipitation: Specific antibodies are employed to identify and bind to RNA fragments possessing specific methylation modifications within cells. For instance, antibodies against m6A modification can recognize and bind to RNA with m6A modification, forming antibody-RNA complexes, which are then bound using magnetic beads and other tools for complex enrichment, thereby enabling the specific separation of methylated RNA from total RNA.
Library construction and sequencing: After the enriched methylated RNA is purified, it is reverse transcribed using a specific kit and method, and a sequencing library is constructed. Subsequently, a large number of short sequence reads are obtained through sequencing on a high-throughput sequencing platform.
Bioinformatics analysis: Quality control and filtering are performed on the original sequencing data, eliminating linker sequences and low-quality bases. The filtered data is then aligned to the reference genome. Owing to the numerous read alignments at methylation modification sites in the sequencing data, "peaks" are formed. By detecting the positions of these peaks, the methylation regions on RNA can be determined, followed by the localization of methylation sites and quantitative analysis of the methylation level, ultimately yielding the map and related information of RNA methylation within the whole transcription group.
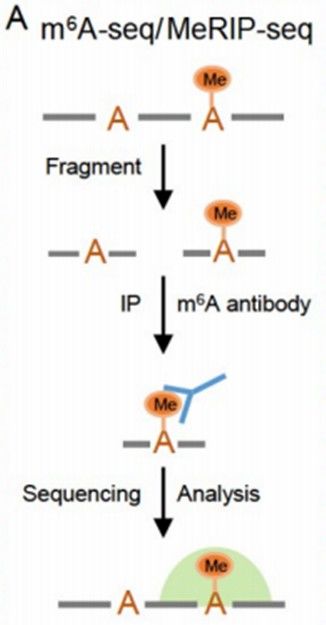 Principle of MeRIP-seq (Yang et al., 2023)
Principle of MeRIP-seq (Yang et al., 2023)
How Does MeRIP-seq Work
MeRIP-seq accurately identifies m6A-modified RNA regions by extracting RNA, selectively enriching m6A-modified RNA, constructing libraries, and performing high-throughput sequencing, providing powerful support for studying RNA modifications and their role in diseases.
Sample preparation: Cell or tissue samples are collected, and total RNA is extracted. Quality inspection is carried out, such as agarose gel electrophoresis to assess the degradation degree and contamination of RNA, which necessitates the presence of distinct 18S or 28S main bands and clear bands. RNA concentration is accurately quantified using Qubit 2.0, with a total amount of not less than 10μg. RNA integrity is precisely detected using Agilent 2100, with an RIN value of not less than 7.5.
Library construction: 10μg of total RNA is taken, and RNA Fragmentation Reagents are added. The reaction is conducted in a Thermomixer at 70℃ for 10 minutes to fragment the RNA into approximately 100nt fragments, which are then precipitated with ethanol. The magnetic beads containing protein A and protein G are washed with IP buffer and incubated with 5μg of m6A antibody at 4℃ for 2 hours. After washing twice with IP buffer, the magnetic beads are resuspended, and the fragmented RNA is added. The mixture is incubated at 4℃ for 4 hours with gentle agitation. Subsequently, the magnetic beads are washed three times with IP buffer at 4℃, incubated with m6A competitive eluent at 4℃ for 1 hour, and the supernatant containing the eluted m6A RNA is collected and purified using phenol chloroform and isoamyl alcohol. IP and Input samples are reverse transcribed and library construction is performed according to the Smarter Stranded Total RNA-Seq Kit V2-Pico Input Mammalian User Manual, and the fragment size is selected using AMPure XP Bead to obtain the final library.
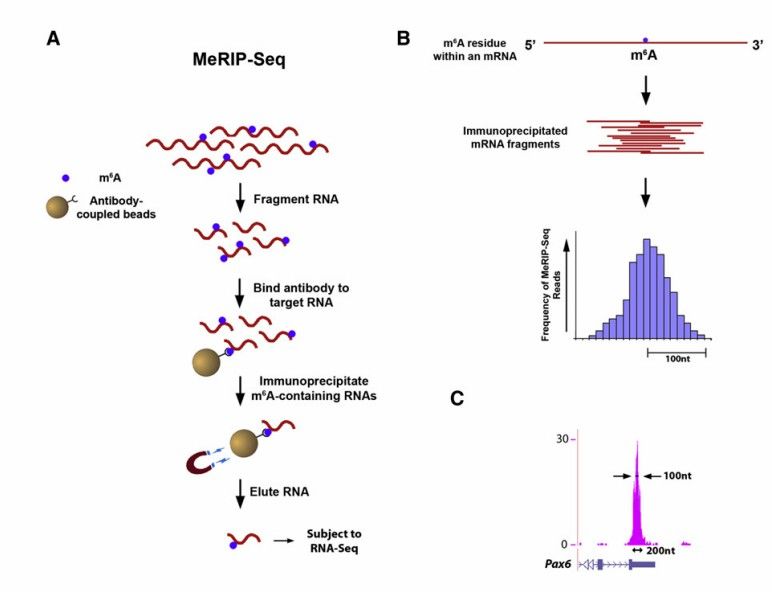 Protocol of Merip-seq (Kate et al., 2012)
Protocol of Merip-seq (Kate et al., 2012)
Library quality inspection: The library is initially quantified using Qubit 2.0 and diluted to 1 ng/μL. Subsequently, the Insert Size of the library is detected using Agilent 2100. Once it meets the expectations, the effective concentration of the library is accurately quantified using the qPCR method to ensure it is greater than 2nM.
Computer sequencing: After the library is qualified, different libraries are pooled and sequenced on the Illumina Nova platform in accordance with the requirements of effective concentration and target offline data, with a sequencing strategy of PE150.
Detection of m6A modified region: Since the number of reads covering the m6A modified region in the IP library is significantly higher than that in the Input library, a peak is formed, and the position of m6A-modified RNA can be determined by detecting the position of the peak.
How to Analyse MeRIP-seq Data
MeRIP-seq data analysis involves quality control, alignment, detection of methylation sites, identification of differential methylation regions, and functional enrichment analysis, providing insights into the role of RNA methylation in biological processes and diseases.
Data preprocessing: Software such as FastQC is used to assess the quality of the original sequencing data, including sequencing base quality, GC content, and sequence length distribution. If the terminal base quality of the sequencing data is found to be low, Trimmomatic software can be employed to remove low-quality bases and linker sequences to ensure the data quality meets the requirements for subsequent analysis. Commonly used tools like HISAT2 or STAR are used to align the filtered data to the reference genome or transcriptome. For example, when studying human samples, the data is aligned to the reference sequence of the human genome to ascertain the origin location of the sequencing reads.
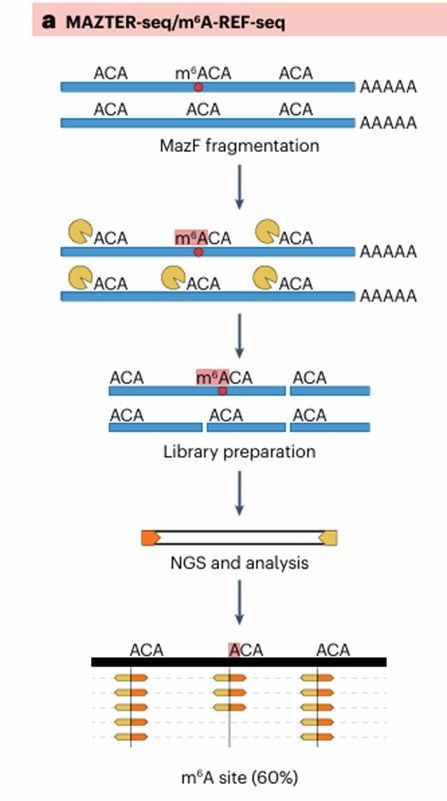 Additional methods for m6A profiling (Michal et al., 2024)
Additional methods for m6A profiling (Michal et al., 2024)
Identification and analysis of methylation sites: Software such as MACS2 is used to detect methylation-rich regions, i.e., peaks, which represent the potential locations of methylation sites. For example, by comparing the sequencing data of immunoprecipitation samples and input samples, the region with a higher enrichment factor is regarded as the potential methylation site region. The methylation level of methylation sites or regions is calculated, generally based on the number of reads covering a specific area in immunoprecipitation samples and input samples to reflect the methylation degree of the site.
Screening differential methylation regions (DMRs): Using software like DiffBind, the methylation levels between different sample groups (such as the disease group and the normal group) are compared, and DMRs are screened out. These DMRs may be associated with the occurrence and development of diseases.
Functional enrichment analysis: The functional enrichment of DMR-related genes is analyzed, often using tools such as DAVID or Metascape to understand the biological processes, molecular functions, and signal pathways involved by these genes. If the related genes are found to be enriched in pathways related to cell proliferation and differentiation, it can be inferred that methylation modification plays a role in these processes.
If you want to learn more about the MeRIP-seq, please refer to:
Advantages of MeRIP-seq
Low initial amount: The sample initial amount can be reduced to 10 - 20μg, with a minimum of only 1μg of total RNA, making it more applicable to some rare samples or those difficult to obtain in large quantities.
Transcriptome-wide detection: It can simultaneously detect mRNA, lncRNA, and other RNAs, enabling the analysis of RNA methylation at the whole transcriptome level, which is beneficial for comprehensively understanding the role of RNA methylation in gene expression regulation.
The sample has strong applicability: It can be used to detect RNA methylation in animals, plants, cells, and tissues, and is not restricted by species, provided that the sample can supply sufficient RNA.
High repeatability: IP enrichment exhibits high repeatability, minimizing the deviation of antibody enrichment, and the experimental results are stable and reliable, facilitating comparison and verification between different samples.
Wide application: It is widely used in many research fields, such as tissue development, self-renewal and differentiation of stem cells, response to heat shock or DNA damage, occurrence and development of cancer, drug response and so on, which is of great significance for in-depth exploration of biological processes and disease mechanisms.
The technical platform is perfect: It offers one-stop service from MeRIP experiment, library construction sequencing to MeRIP-qPCR verification, and has a variety of library construction methods and fragment sizes to choose from, providing flexibility to meet different scientific research requirements.
Accurate quantification: Methylated fragments can be directly quantified and sequenced, avoiding the cross-reaction and background noise of traditional chip hybridization fluorescence analog signals, resulting in more accurate and reliable detection results.
Limitations of MeRIP-seq
Effect of antibody specificity: The accuracy of test results is highly reliant on the specificity and affinity of antibodies. If the antibody performance is suboptimal, it may lead to nonspecific enrichment and affect the accurate determination and quantitative analysis of methylation loci.
Limited resolution: Although the methylation site can be located within the transcriptome, it can only be pinpointed to approximately 100 - 200 nucleotides, making it challenging to identify the methylation site at a single nucleotide level, and thus presenting limitations in some studies requiring high-precision methylation information.
Inability to distinguish different methylation states: It can only detect whether RNA is methylated or not, and cannot discriminate between different degrees of methylation modification at the same methylation site, such as monomethylation and bimethylation, which restricts in-depth research on the complexity of methylation modification.
Sample bias exists: Under different batches of samples or experimental conditions, the results may be skewed due to factors such as sample processing and immunoprecipitation efficiency, which will impact the comparability and repeatability of the data. Stringent control of experimental conditions and multiple repetitions of experiments are necessary.
Complex data analysis: It entails extensive bioinformatics analysis, including methylation site identification, peak detection, difference analysis, etc, demanding high professional skills and computing resources of analysts, and the interpretation of analysis results also requires abundant experience and professional knowledge.
Influencing Factors of MeRIP-seq
MeRIP-seq results can be influenced by factors such as RNA integrity, purity, sample size, antibody performance, and crosslinking efficiency, all of which must be carefully optimized to ensure accurate and reliable data.
RNA integrity: If RNA is degraded, methylation site information will be lost or false negative results will occur. Generally, the RIN value of RNA is required to be no less than 7.0.
Purity of RNA: The presence of impurities such as protein and DNA will interfere with the binding between antibody and RNA and subsequent experimental steps, affecting the accuracy of the results.
Initial amount: An insufficient sample size makes it difficult to obtain an adequate amount of methylated RNA fragments, while an excessive sample size may lead to increased nonspecific binding. Generally, the initial amount of total RNA is not less than 10μg.
Antibody performance: If the antibody fails to accurately recognize the modified base corresponding to the target methylation site, it will result in nonspecific enrichment and produce false positive results. Low affinity will lead to insufficient enrichment efficiency, affecting the detection of low-abundance methylation sites and leading to inaccurate results.
Crosslinking efficiency: Before immunoprecipitation, if the dosage of the cross-linking agent, cross-linking time, and other conditions are inappropriate, the cross-linking between RNA and protein will be incomplete, leading to a decrease in the precipitation efficiency of methylated RNA and affecting the detection results.
References:
- Yang, Wenlan, Yongliang Zhao, and Yungui Yang. "Dynamic RNA methylation modifications and their regulatory role in mammalian development and diseases." Science China Life Sciences (2024): 1-21. https://doi.org/10.1007/s11427-023-2526-2.
- Yadav, Pooja, et al. "M6A RNA methylation regulates histone ubiquitination to support cancer growth and progression." Cancer research 82.10 (2022): 1872-1889. DOI: 10.1158/0008-5472.CAN-21-2106.
- Moshitch-Moshkovitz, Sharon, et al. "mRNA m6A detection." Nature Reviews Methods Primers 4.1 (2024): 1-20. https://doi.org/10.1038/s43586-024-00365-9
! For research purposes only, not intended for clinical
diagnosis, treatment, or individual health assessments.





