
RNA modification refers to various chemical modifications (over 160 types) that occur on RNA molecules. Each modification has specific functions, such as stabilizing structure, nuclear export, alternative splicing, translation recognition, and more. Methylation is one of the major forms of RNA modification, accounting for approximately two-thirds of all modifications. RNA methylation can occur on mRNA, tRNA, rRNA, and other non-coding RNAs. Among them, m7G modification (N7-methyladenosine) is a positively charged RNA methylation modification. Under the action of methyltransferases, a methyl group is added to the seventh nitrogen atom of RNA guanine (G), resulting in this chemical modification. m7G RNA methylation is commonly found in the 5' cap structure of eukaryotic mRNA and plays a role in regulating mRNA transport, translation, and splicing processes. In addition to the 5' cap structure, m7G modification also exists within tRNA and rRNA sequences. With the widespread application of NGS, RNA modification has become a hot topic in the field of epigenetics, and MeRIP-seq is a powerful technique for studying m7G modification.
MeRIP-seq combines MeDIP (methylated DNA immunoprecipitation), RIP (RNA immunoprecipitation), and RNA-seq technologies to accurately detect RNA methylation at the genome-wide level. It involves fragmenting RNA samples and performing immunoprecipitation using specific antibodies against RNA methylation, followed by sequencing of the methylated fragments. At the same time, an input control sample (similar to ChIP-seq) is designed to eliminate background noise.
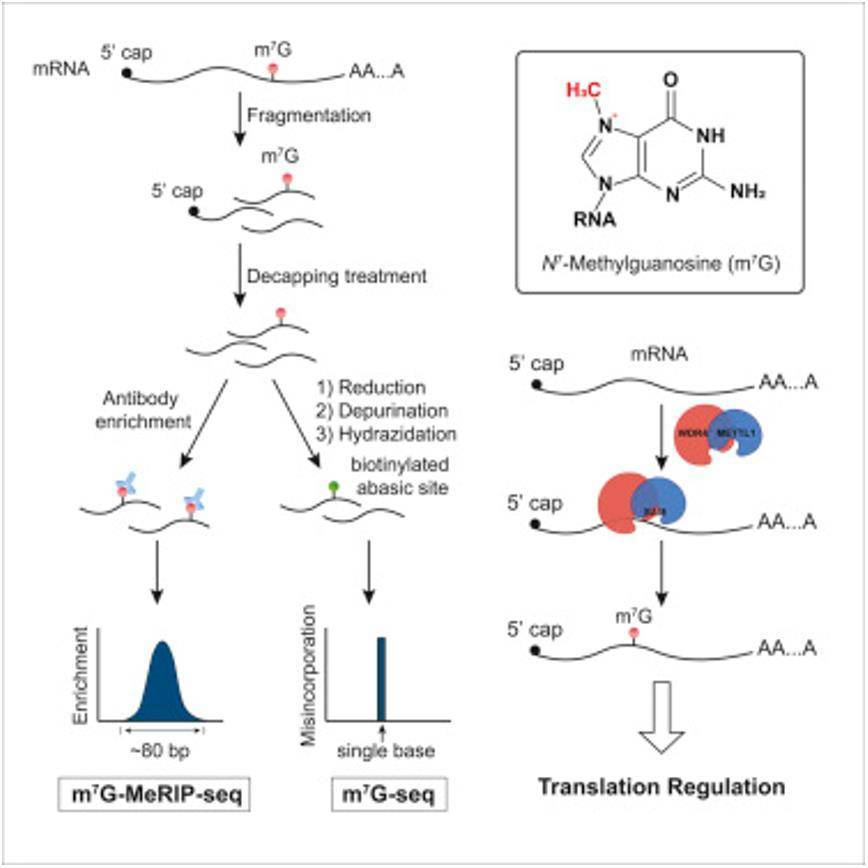
m7G-meRIP-seq Wokflow
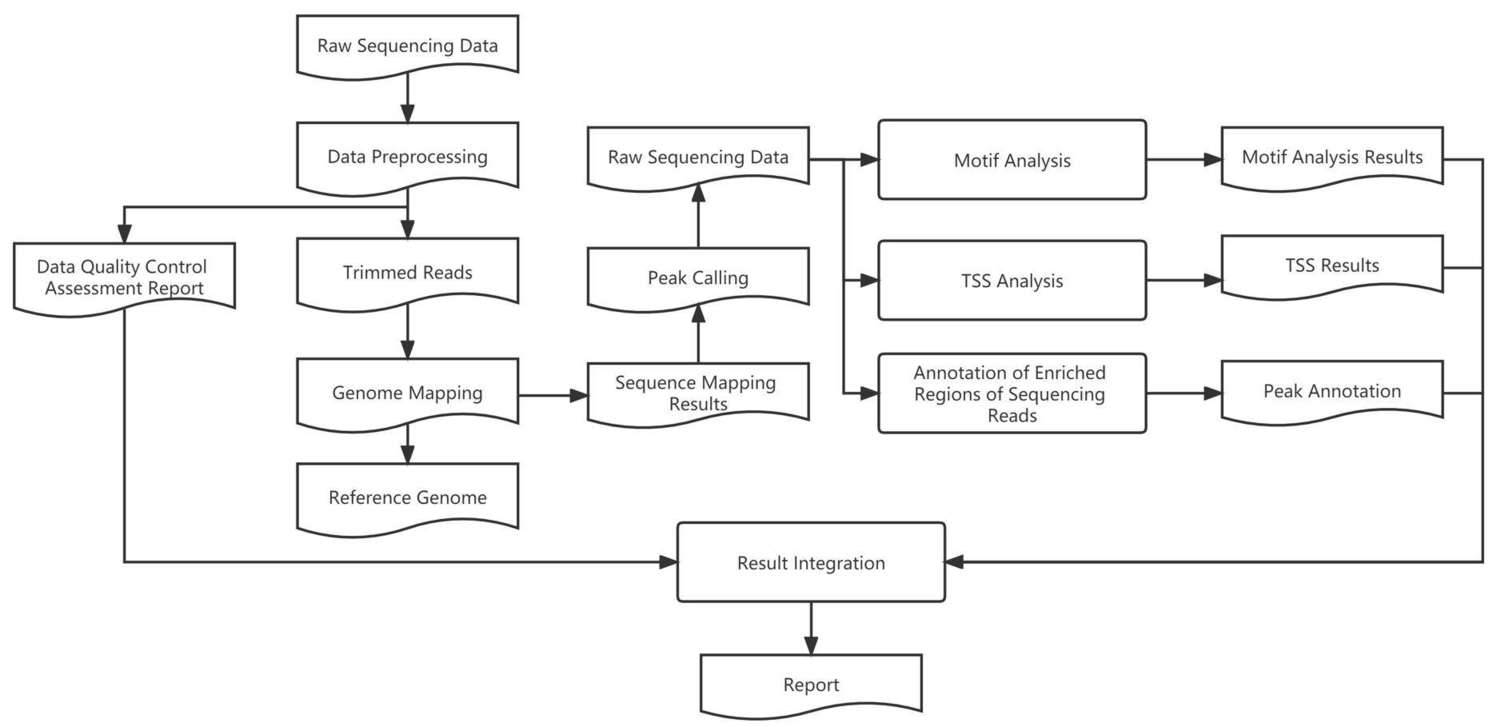
Library Construction:
The construction of MeRIP-seq libraries involves the following steps:
Total RNA extraction and removal of DNA contamination using DNase I.
RNA precipitation and ethanol washing, followed by resuspension in ultrapure water.
Measurement of RNA concentration and fragmentation of RNA using a thermal cycler.
Evaluation of the size of fragmented RNA (around 200 nt).
Incubation with anti-m7G antibody.
Wash and elution to enrich the m7G-modified RNA.
Construction of cDNA libraries.
Sequencing on the sequencing platform.
Analysis Workflow:
After obtaining the sequencing data, the analysis workflow is as follows:
Quality assessment of the raw data using FastQC software.
Alignment of clean data to the reference genome using BWA software.
Processing of SAM files using Samtools, including sorting, deduplication, and organization.
Peak calling using MACS2 (q = 0.01) to identify regions of high coverage in the sequencing reads.
Annotation of the identified peaks and motif analysis using Deeptools.
Utilization of the R language for data statistics, plotting, differential analysis, and functional enrichment analysis.
m7G-meRIP-seq Technical Advantages
Low input requirement: Sample input can be reduced to 10-20 μg, with a minimum requirement of only 1 μg total RNA.
High-throughput sequencing: Enables high-throughput sequencing of m7G sites in the entire transcriptome, allowing simultaneous detection of mRNA and lncRNA.
Broad sample applicability: Suitable for m7G detection in animals, plants, cells, and tissues.
High reproducibility: m7G-MeRIP-seq demonstrates high reproducibility in IP enrichment, minimizing antibody enrichment bias.
Wide range of applications: Widely used in research areas such as tissue development, stem cell self-renewal and differentiation, response to heat shock or DNA damage, cancer initiation and progression, drug response, and more.
Sample Requirements
RNA Sample: Total RNA: ≥300 μg, with an RNA Integrity Number (RIN) value greater than 7.5.
Cells or Tissues: Cell Quantity: ≥30 million cells Animal Tissue Quantity: ≥500 mg Plant Tissue Quantity: ≥5 g
It is recommended to prepare 2-3 replicates of the sample. For detailed information on sample preparation, please contact us.
Transcriptome-wide Mapping of Internal N7-Methylguanosine Methylome in Mammalian mRNA
Journal: Molecular Cell
Impact Factor: 14.25
In order to accurately investigate the internal m7G methylation in RNA, which cannot be reverse transcribed by any known reverse transcriptase into cDNA with corresponding base mutations, the team led by Chuan He developed a novel deep sequencing method based on the inherent positive charge of m7G. This method converts m7G sites in RNA into new sites that can undergo reverse transcription base mutations, and estimates the methylation level of m7G sites based on the mutation rate. Subsequently, this method was proven to detect 1639 internal m7G sites in 18S rRNA and revealed 22 internal m7G sites in human cellular tRNA. The distribution, enrichment of common sequences, and other statistical features of m7G revealed by this method were consistent with the m7G-MeRIP-seq data. This study not only elucidated the distribution characteristics of m7G methylation in human cells but also identified METTL1 as a methyltransferase that catalyzes m7G methylation in mRNA, indicating that internal methylation of m7G can affect mRNA translation.
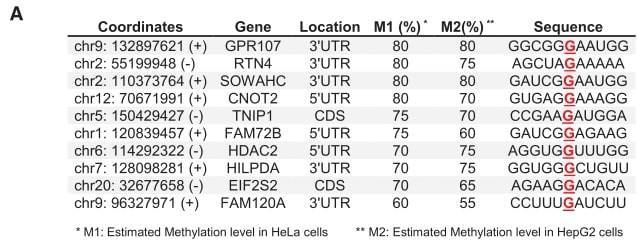
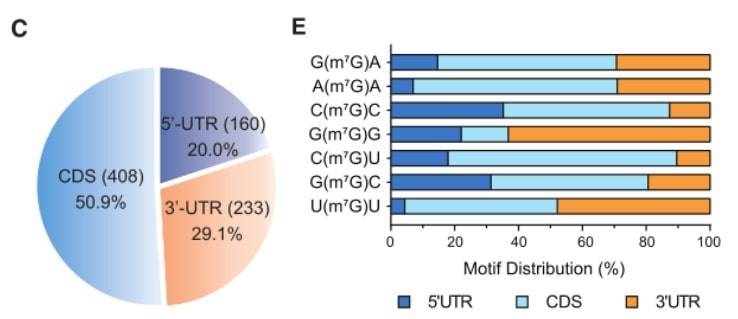 (A) m7G single-base deep sequencing was performed on Hela and HepG2 cells, and 10 representative mRNA internal m7G modification results are shown. (C) mRNA characteristics of 801 m7G sites. (E) Distribution percentages of the top 7 mRNA internal m7G motifs in mRNA.
(A) m7G single-base deep sequencing was performed on Hela and HepG2 cells, and 10 representative mRNA internal m7G modification results are shown. (C) mRNA characteristics of 801 m7G sites. (E) Distribution percentages of the top 7 mRNA internal m7G motifs in mRNA.
Reference
- Li-Sheng Zhang et al,. Transcriptome-wide Mapping of Internal N7-Methylguanosine Methylome in Mammalian mRNA. Molecular Cell 2019