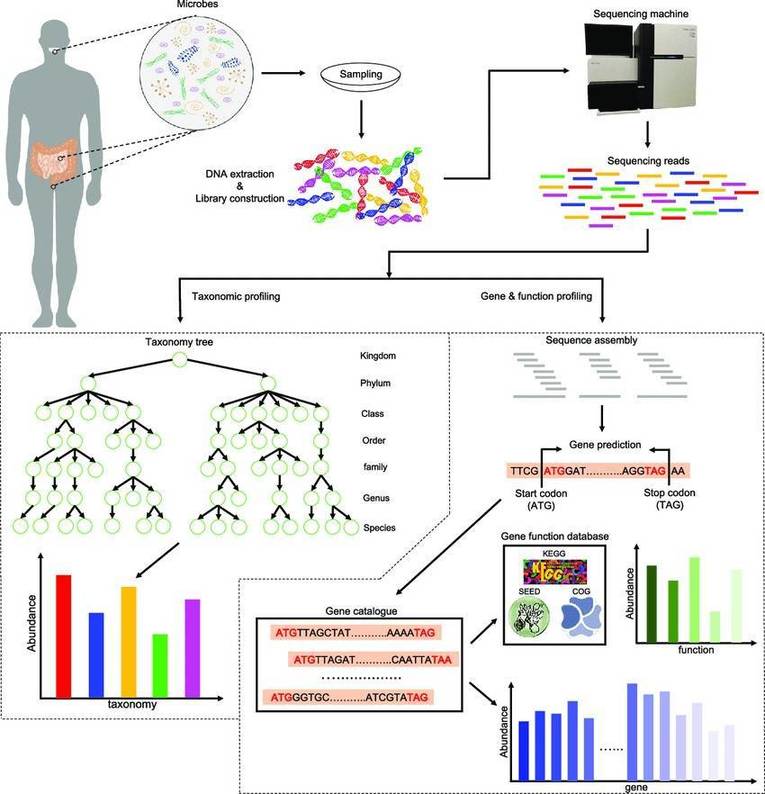
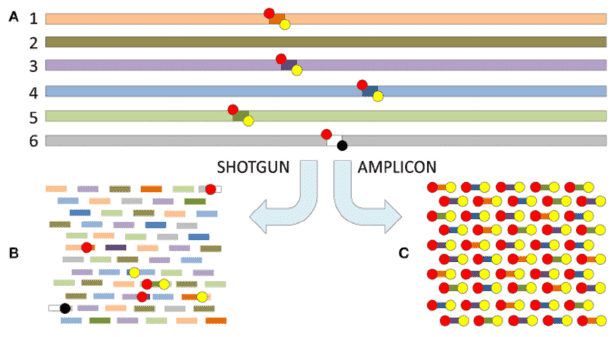
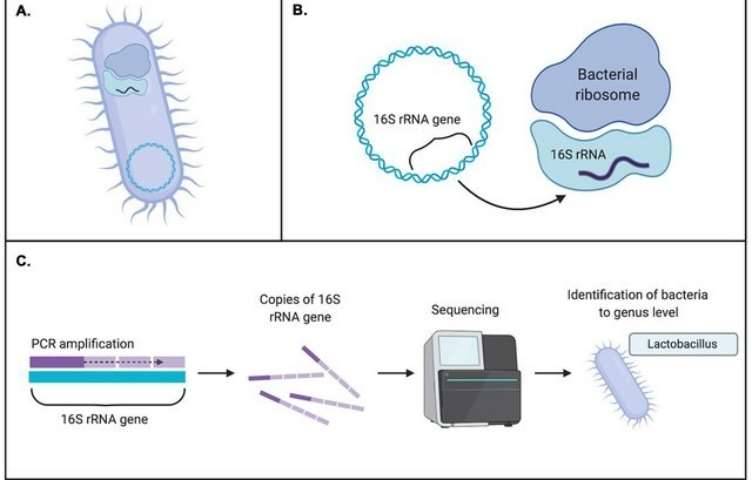
The application of microbiomics is now widespread across various research domains, including studies centered on gut microbiota, food fermentation, plant rhizosphere microbiota, and environmental microbiota, among others. Within this context, the usage of various metagenomic databases plays an indispensable role. This article provides a concise summary of frequently utilized databases in the realm of Metagenome analysis.
Which Database for Metagenome Annotation to Chose?
The KEGG Database
The Kyoto Encyclopedia of Genes and Genomes (KEGG) is an online repository pertaining to genomes, enzymatic pathways, and biochemical substances. The records within the PATHWAY database depict interactions amongst various molecules within cells, alongside unique physiological alterations encountered within specific organisms. The KEGG database frequently stands out as the go-to resource across various '-omics' platforms, serving as a critical tool for unraveling the underlying functional mechanisms within microbial communities.
COG Database
The Cluster of Orthologous Groups of proteins (COG) is a database that classifies the systematics and evolutionary relationships of encoded proteins in organisms with complete genomes, including bacteria, algae, and eukaryotes. COG comprises clusters of protein orthologs and paralogs, each assumed to stem from an ancestral protein. Orthologs, proteins from separate species that have descended from a common ancestor, tend to retain identical functions to the original protein. Conversely, paralogs, which result from gene duplication, often evolve to perform varied functions than the ancestral protein. The primary application of COG lies in identifying orthologous genes in newly sequenced genomes and offering function predictions for these identified genes at a high resolution.
eggNOG Database
The eggNOG database, an acronym for the Evolutionary genealogy of genes: Non-supervised Orthologous Groups, is dedicated to providing functional descriptions and classification annotations for orthologous gene clusters. This database enables a comprehensive understanding of microbial community gene functions and their evolutionary relationships.
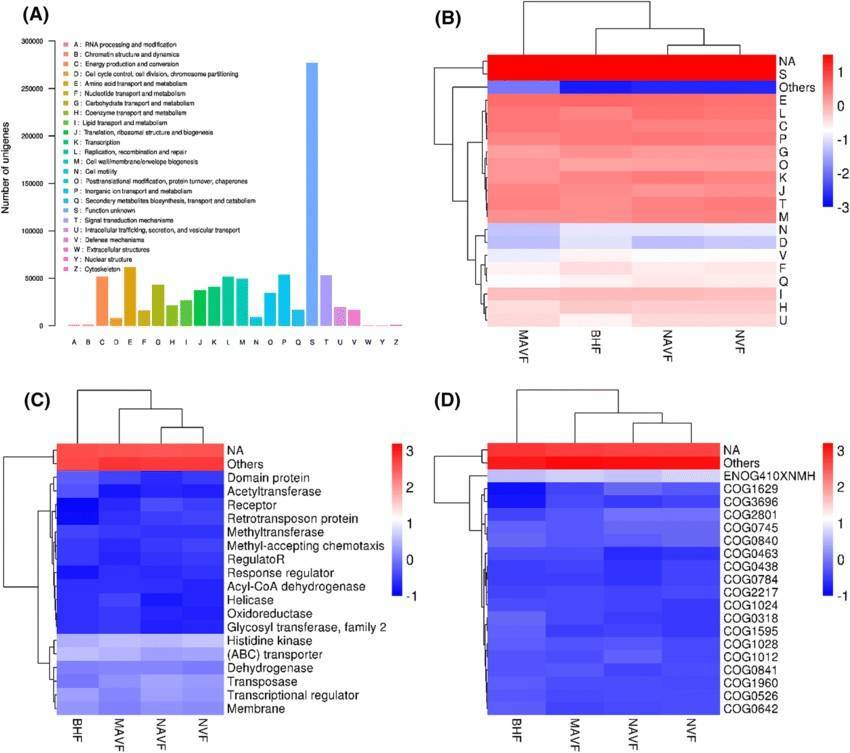 eggNOG annotation (Shiyang Zhang et al,. 2020)
eggNOG annotation (Shiyang Zhang et al,. 2020)
CAZy Database
The CAZy database, fully termed as Carbohydrate-Active enZymes, is an authoritative database centred around enzymes involved in carbohydrate metabolism. The database operates on the basis of sequence similarity, classifying enzymatic sequences into five major categories, namely: glycoside hydrolases (GHs), glycosyltransferases (GTs), polysaccharide lyases (PLs), carbohydrate esterases (Ces), and carbohydrate-binding modules (CBMs). The CAZy database has a profound association with cell wall degradation, hence it is frequently employed in the exploration of pathogenic mechanisms of plant pathogens.
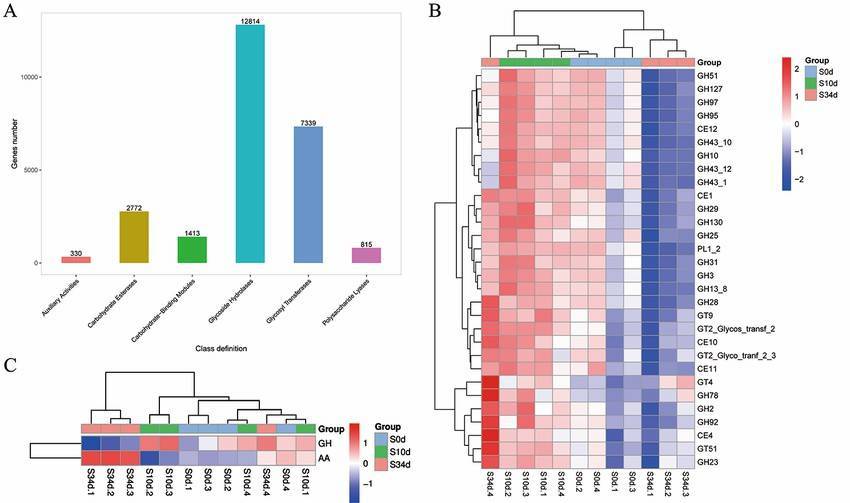 CAZyDatabase Annotation (Huan Xu et al,. 2022)
CAZyDatabase Annotation (Huan Xu et al,. 2022)
CARD Database
The Comprehensive Antibiotic Resistance Database, abbreviated as CARD, is dedicated to providing data, models, and algorithms related to antibiotic resistance on the molecular level. The core of this database is the Antibiotic Resistance Ontology (ARO), which aims for a thorough and systematic description of antibiotic resistance. Plasmids, commonly found in pathogenic bacteria, often carry a vast number of resistance genes and are closely associated with horizontal gene transfer events. The information detailed in CARD is crucial for deepening our understanding of the resistance mechanisms utilized by pathogenic bacteria, as well as for the prevention and control of resistance transmission.
Swissprot Database
The Swiss-Prot database, a manually annotated protein sequence repository, is maintained by the European Bioinformatics Institute (EBI). This database primarily encompasses protein sequence entries, each of which includes sequence information, referenced bibliographic details, taxonomic data, and accompanying annotations. The annotation section provides comprehensive details pertaining to the functionality of the protein, its post-translational modifications, specialized spots and zones, and also incorporates descriptions of secondary and quaternary structures. Additionally, it encompasses information such as homology with other sequences, associations between sequence remnants and diseases, variations in sequences, conflicts, and more.
PHI Database
The PHI-base, or the Pathogen-Host Interactions database is dedicated to analyzing the interplay between plant or animal pathogens and their hosts to elucidate the pathogenic mechanisms that exist between them.
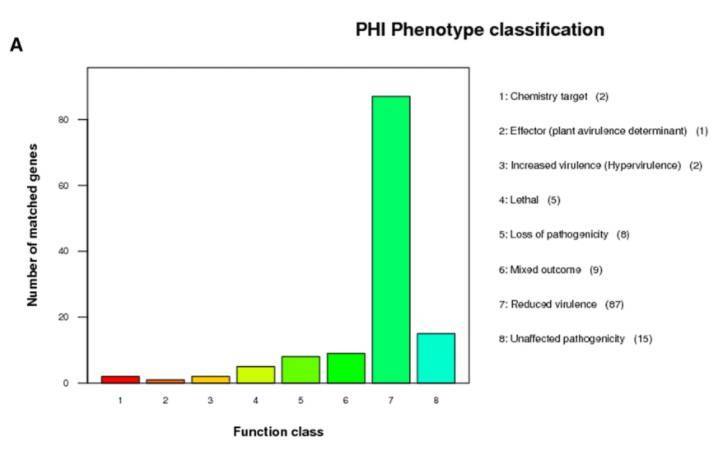 Annotation based on the PHI database (Bowen Wang et al,. 2019)
Annotation based on the PHI database (Bowen Wang et al,. 2019)
Mobile Genetic Element Database
The Mobile Genetic Elements (MGE) database, referred to as the MGE database, is a reference gene database incorporating a wealth of mobile genetic elements. Currently, this database has collated 278 unique genes alongside over 2000 reference sequences, encompassing an array of mobile genetic elements such as Insertion Sequences (IS), transposons, integrons, self-transmissible broad host range plasmids, genomic islands, and phages. Mobile Genetic Elements (MGEs) denote gene assembly fragments equipped with specific functional genes, capable of moving freely within and outside cells. These mobile elements play a pivotal role in bacterial horizontal gene transfer, genome recombination, and species evolution.
BacMet Database
The BacMet database, officially known as the Antibiotic and Heavy Metal Resistance Genes Database, is a specialized and authoritative resource containing 753 genes associated with resistance to heavy metal ions and biocides. Leveraging this database, researchers are able not only to identify genes conferring resistance to heavy metal ions and biocides in the target strains, but also to accurately determine genes rendering dual resistance. This unique feature underscores the irreplaceable importance of the BacMet database in the realm of strain resistance research.
The unique feature of this database is its function-based gene selection process, meticulously categorizing metal resistance genes and antibacterial resistance genes according to their resistance types and genealogical relationships. Such taxonomic approach holds significant guidance for data mining in environmental microbiological communities, such as various types of soil microbial communities, and anaerobic fermentation of activated sludge, among others.
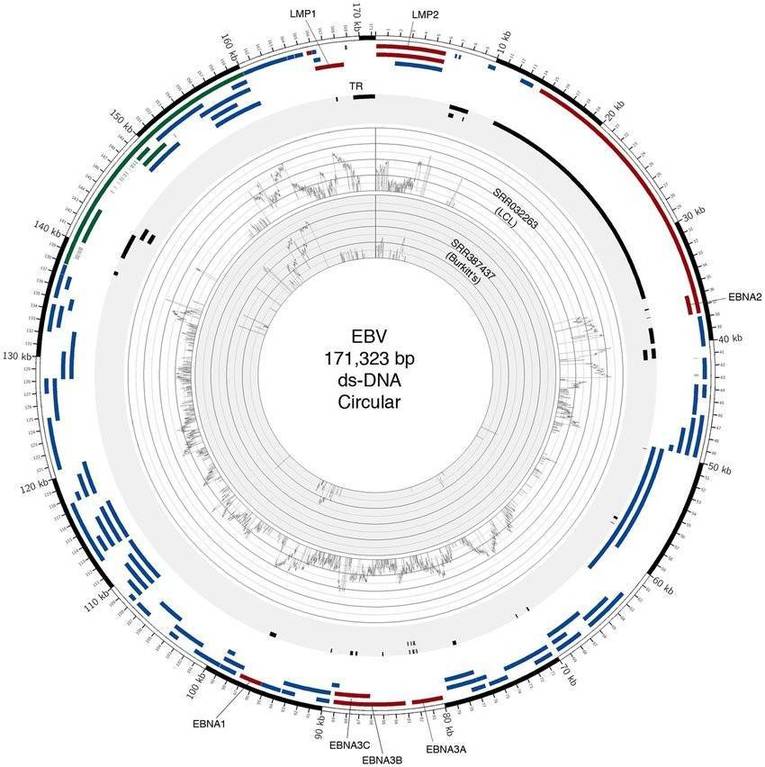 Circos plot of two EBV (Guorong Xu et al,. 2014)
Circos plot of two EBV (Guorong Xu et al,. 2014)
UHGG Database
The Unified Human Gastrointestinal Genome (UHGG) collection comprises 204,938 non-redundant genomes derived from 4,644 gut prokaryotic species, along with over 170 million protein sequences. By employing the UHGG database in association with Kraken2 for alignment, we can more comprehensively elucidate the taxonomic diversity of the human gut microbiota.
How to Annotate Metagenome Databases?
Analyzing Phenotypic Features of Specific Samples:
In microbial communities, notable differences in phenotypic characteristics among diverse comparative groups indicate significant variations in both the overall species distribution and functionality. These variations play a crucial role in preserving the equilibrium of the microbial ecosystem. Given these dynamics, when collecting microbial community samples for scientific inquiry, it becomes imperative to meticulously gather all relevant phenotypic data linked to the study. This meticulous approach not only offers a comprehensive perspective for subsequent data mining activities but also enhances the depth of scientific comprehension and interpretation.
Integrating analyses with other omics datasets
Exploring the intricate association between the focal species and the regulatory mechanisms steering community dynamics necessitates a thorough analysis. This can be achieved through an integrated approach that amalgamates transcriptomic, metabolomic, human resequencing, single-cell transcriptomic data, and findings derived from metagenomic analysis. The collective body of omics studies consistently reveals a direct interrelation between microbial communities and microglial cells within the brain, furnishing compelling evidence to advance our comprehension of the mechanisms orchestrating their interactions.
Unveiling the functions associated with core genes and species
Within microbial co-occurrence networks, core genes and species typically occupy pivotal nodal positions, potentially attributed to their high abundance or specific functionalities. Simultaneously, certain low-abundance microbial pathogens may influence the structure and functionality of the entire microbial community by altering the normal interplay of microbial consortia. To gain profound insights into the mechanisms governing microbial communities, an in-depth exploration and understanding of these microbes can be achieved through the analysis of core genes and species. This involves exploring relevant database annotations to further investigate and comprehend these microbial entities.