




MeRIP-seq: Principle, Protocol, Bioinformatics and Applications in Diseases
Epitranscriptome pertains to the phenomenon wherein gene expression is modulated by chemical alterations on RNA without altering the RNA sequence. There exist over 100 types of intracellular RNA modifications, with the majority being conspicuous modifications on tRNA and other non-coding RNA, and only a few modifications on mRNA. The most abundant modification in mRNA is N6-methyladenosine (m6A), which partakes in all facets of the mRNA life cycle. Research has demonstrated that the primary function of m6A is to govern the stability of mRNA.
Detection Theory of MeRIP-seq
Immunocoprecipitation: Specific antibodies are employed to identify and bind to RNA fragments possessing specific methylation modifications within cells. For instance, antibodies against m6A modification can recognize and bind to RNA with m6A modification, forming antibody-RNA complexes, which are then bound using magnetic beads and other tools for complex enrichment, thereby enabling the specific separation of methylated RNA from total RNA.
Library construction and sequencing: After the enriched methylated RNA is purified, it is reverse transcribed using a specific kit and method, and a sequencing library is constructed. Subsequently, a large number of short sequence reads are obtained through sequencing on a high-throughput sequencing platform.
Bioinformatics analysis: Quality control and filtering are performed on the original sequencing data, eliminating linker sequences and low-quality bases. The filtered data is then aligned to the reference genome. Owing to the numerous read alignments at methylation modification sites in the sequencing data, "peaks" are formed. By detecting the positions of these peaks, the methylation regions on RNA can be determined, followed by the localization of methylation sites and quantitative analysis of the methylation level, ultimately yielding the map and related information of RNA methylation within the whole transcription group.
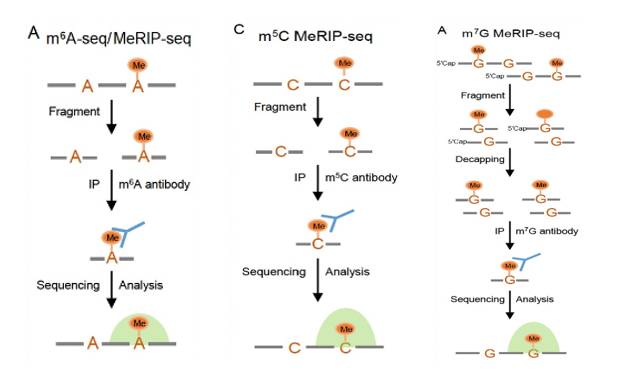 Principle of MeRIP-seq (Yang et al., 2023)
Principle of MeRIP-seq (Yang et al., 2023)
MeRIP-seq Operational Workflow
Methylated RNA-specific antibodies are incubated with randomly interrupted RNA fragments, and methylated fragments are grabbed for sequencing. At the same time, a control sample is sequenced in parallel, and the sequence fragments in the immunoprecipitation sample and the control sample are mapped to the reference genome or transcription group, so as to detect RNA methylation sites.
Sample preparation
RNA extraction: To extract high-quality total RNA from cells, tissues and other samples, it is required that RNA has good integrity and no obvious degradation. Generally, 2× 10 cells, 100mg-1g tissues and 30-300μg RNA are needed.
Fragmentation: randomly interrupt the extracted RNA to make it into a moderate length fragment, generally about 100-200 nucleotides.
Immunocoprecipitation
Incubation of antibodies: The fragmented RNA is co-incubated with antibodies with specific methylation modifications, such as m6A specific antibodies, under appropriate conditions, so that the antibodies can specifically bind to RNA fragments with methylation modifications.
Capture methylated fragments: Use protein A/G magnetic beads and other tools to capture antibody-RNA complex, thus enriching RNA fragments containing methylation modification.
Library construction
Elution and purification: the captured methylated RNA fragments were eluted from the magnetic beads and purified to obtain pure methylated RNA.
Reverse transcription and amplification: using purified methylated RNA as a template, reverse transcription is carried out to synthesize cDNA, and then sequencing library is constructed by PCR amplification and other means.
High throughput sequencing
Choose sequencing platform, such as Illumina NovaSeq, and sequence the constructed library on computer to obtain a large number of sequencing readings.
Data preprocessing: Filter the original sequencing data to remove linker sequence, low-quality reading length and pollution sequence, and get high-quality clean data.
Comparison and localization: Compare clean data with reference genome or transcriptome to determine the position of sequencing reading length on genome.
Peak identification and annotation: Peak calling is carried out by bioinformatics algorithm to identify methylation-rich regions, and these regions are annotated to determine their positions in gene elements.
Differential analysis: Compare methylation levels among different samples, identify differential methylation sites or regions, and further analyze the relationship between them and gene expression and phenotype.
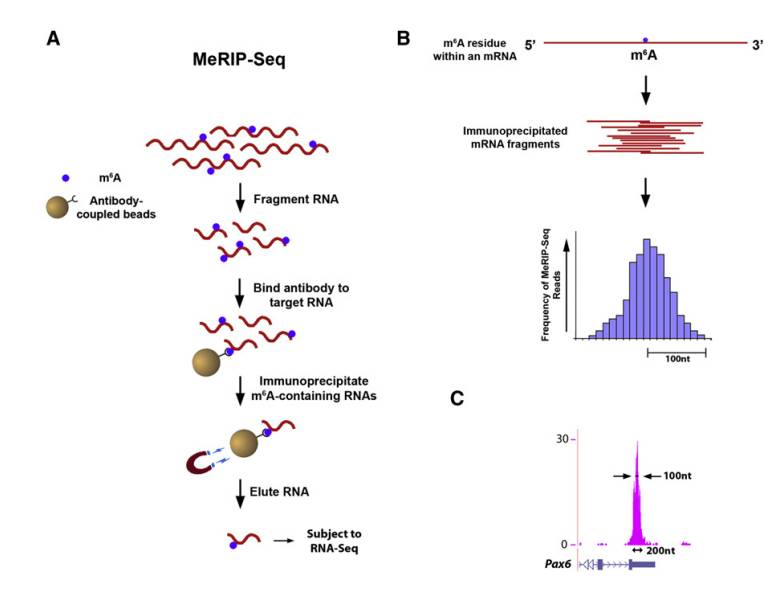 Protocol of Merip-seq (Kate et al., 2012)
Protocol of Merip-seq (Kate et al., 2012)
Service you may intersted in
If you want to learn more about the MeRIP-seq, please refer to:
Bioinformatics Analysis of MeRIP-seq
Merip-seq biological analysis is aimed at RNA methylation. At first, the data is preprocessed to eliminate low-quality sequences and joint pollution and purify the data. Subsequently, the sequence is aligned to the reference genome to locate the location of RNA. Then the methylation sites are identified and their levels are quantified. By comparing different samples, the differential methylation regions are mined and the key methylation modifications are identified. Functional annotation and enrichment analysis revealed the function and participation pathway of methylation-related genes. It can also integrate other omics data to comprehensively analyze the role of methylation in gene expression regulation and biological phenotype, and provide key insights for further understanding the biological mechanism of RNA methylation.
Data preprocessing
Remove connectors and low-quality reads: Use Cutadapt, Trim Galore! And other tools to remove sequencing linker sequences, prune low-quality bases and improve data quality.
Compare with reference genome: compare high-quality reads with HISAT2, STAR, etc. to the reference genome, and determine its position on the genome.
Methylation site recognition
Peak detection: use MACS2 and other software to identify the enrichment area in MeRIP-seq data, that is, the methylation peak, which represents the possible RNA methylation site.
Annotation peak: Use bedtools and other tools to associate the peak with gene structure annotation file (GTF/GFF) to determine methylation.locusAnd gene regions (promoter, coding area, UTR, etc.).
Differential methylation analysis
Sample grouping: Group the samples according to the experimental purpose (disease group and control group).
Identification of Differential Methylation Region (DMR): Use DSS and other software to analyze the difference of methylation level between groups and determine DMR.
Functional enrichment analysis: For DMR-related genes, DAVID and Metascape were used to do GO and KEGG enrichment analysis to understand the potential functional impact of differential methylation.
Association analysis with gene expression
Obtain gene expression data: it can come from the same batch of samples RNA-seq, or public database.
Correlation analysis: using statistical methods such as Spearman correlation analysis, the relationship between methylation level and gene expression was studied, and the regulatory effect of methylation on gene expression was judged.
Results visualization
With the help of R language, Python and other programming languages and related drawing packages, or using the visualization function of tools such as MeRIPSeqPipe, the analysis results are displayed in the form of peak distribution density curve, motif detection result map, differential methylation heat map, volcano map and differential expression gene heat map, so as to present data characteristics and analysis results more intuitively.
DMR Analysis of MeRIP-seq
DMR analysis is very important in MeRIP-seq analysis. By comparing different samples, it finds out the regions with significant differences in methylation levels. First, strict data screening and standardization are carried out to ensure reliable and comparable data. Then use special algorithms, such as edgeR and DESeq2, to identify DMR statistically.
After finding DMR, the related genes are annotated and analyzed, and the role of genes in biological processes, cell components and signal pathways is clarified by GO and KEGG enrichment analysis. These analyses can reveal the differences of methylation patterns among samples, help to understand the regulatory mechanism of methylation modification in development and diseases, and provide key clues for biomedical research.
Data preparation
Collect methylation sequencing or chip data, ensure data quality, and pre-process, such as removing low-quality data, standardization, etc., to reduce batch effect and other interference factors.
Identification of differential methylation sites
Professional software or tools, such as minfi package, are used to analyze the pretreated data to determine the sites with significant differences in methylation levels between the experimental group and the control group, and the significance of the differences is usually measured by statistics such as P value.
Identification of DMR
Based on the identified differential methylation sites, the adjacent sites are clustered into regions according to the conditions such as distance, which are used as potential DMR. Metilene is a commonly used software, which uses binary separation algorithm combined with double statistical test to determine DMR according to the rules of site sequencing depth and methylation difference.
Notes of DMR
By comparing the identified DMR with the genome annotation file, we can determine its position in the genome, such as the promoter, coding region, UTR and other regions of the gene, clarify its relationship with the gene structure, and understand its potential impact on gene expression.
Screening and evaluation of differentially methylated regions
According to the P value and FDR of statistical test, the DMR was screened, and the areas with significant differences and high reliability were retained. The importance of DMR could be evaluated by combining the length of DMR and the number of CpG sites contained.
Functional enrichment analysis
GO, KEGG and other enrichment analysis were carried out on the genes related to DMR, so as to understand the biological process, molecular function and signal pathway of these genes, and explore the role of DMR in biological function and disease occurrence and development.
Visual display
Using tools such as genome browser, DMR and its related information, such as methylation level change and gene location, are displayed visually, and the distribution and characteristics of DMR in the genome are presented intuitively, which is helpful for analysis and interpretation of the results.
Service you may intersted in
Software for DMR Analysis
DMR analysis is used to identify the different regions of methylation levels among different samples and reveal the role of methylation in biological processes. Some commonly used softwares for DMR analysis are summarized as follows.
Software based on R language
Methylkit: It can process sequencing data such as RRBS and WGBS, accurately regress by Fisher when there is no biological duplication, screen differential methylation sites and regions by logistic regression when there is duplication, and annotate and visualize the data, with rich visualization effect.
DSS: It relies on bsseq package, and its core is a program based on hierarchical Bayesian model, which is used to estimate and reduce the specific dispersion of genes or CpG sites, and then the differential methylation is detected by Wald test, which can analyze the differential expression of RNA-seq and the differential methylation of bisulfite sequencing.
Dmrseq: Based on bsseq package, it mainly detects differential methylation regions or CpGs, and selects them by GLS regression model.
DMRcate: Mainly aiming at Illumina HM450K BeadChip data, Gaussian kernel function is used to spatially smooth the methylation measurement values to identify and rank DMRs, which is not affected by genome annotation or the changing direction of differential methylation signals, and provides statistical significance for each DMR.
PCBS: Differential methylation sites and regions are identified by principal component analysis, and a new algorithm is adopted to select seed sites and expand surrounding regions to identify DMR, which has high computational efficiency and accuracy.
Other software
Nf-core/methylseq: Methylation sequencing data analysis process based on Nextflow platform. Bismark or bwa-meth/MethylDackel can be selected as the core comparison and analysis engine, which is suitable for cancer research and other fields.
Metilene: DMR was determined by binary separation algorithm combined with double statistical test according to site sequencing depth and methylation difference.
Methylasso: Using generalized additive model and fused lasso regression framework, DNA methylation data can be directly modeled without data boxing, and DMR under different conditions can be effectively identified, which is excellent in benchmark test.
Applications of MeRIP-seq
MeRIP-seq is widely used in medical research, including disease diagnosis, disease mechanism research, drug research and development, disease prognosis evaluation, etc. The following are some main application cases.
Provides New Potential Targets
One paper focuses on the effect of long-chain non-coding RNA (lncRNA) methylation in gastric cancer stem cells (GCSCs) on cell apoptosis and dryness, and reveals the key role and potential mechanism of lncRNA methylation in the occurrence and development of gastric cancer, which provides new potential targets and theoretical basis for the treatment of gastric cancer.
MeRIP-seq, quantitative real-time polymerase chain reaction (qPCR), Western blot, single-base extension and ligation qPCR amplification (SELECT), double luciferase reporter gene detection, RNA pull-down experiment and other techniques were used to study the m6A modification level, stability, interaction with lncRNAs and protein, and its influence on cell function. At the same time, the tumorigenesis experiment in vivo was carried out to observe the effect of methylated lncRNAs on the tumorigenicity of GCSCs.
Through the analysis of GCSCs and GCNSCs, it was found that m6A levels of various lncRNAs in GCSCs were increased, among which PAPPA-AS1, PSMA3-AS1, MIR22HG, LINC00342, LINC01410 and LINC00680 were significantly different, and the expressions of PSMA3-AS1 and MIR22HG were up-regulated in GCSCs.
It is determined that METTTL 3 and METTL14 are the key enzymes for methylation of lncRNA in GCSCs, and knocking them down will reduce the m6A level of LNC RNA and the dryness of GCSCs. Site-specific m6A modification of PSMA3-AS1 and MIR22HG can promote the proliferation, inhibit apoptosis and enhance dryness of GCSCs, and m6A modification can enhance the stability of these two lncRNAs.
The methylation of PSMA3-AS1 and MIR22HG is mediated by METTL3 and METTL14 in GCSCs, and its site-specific m6A modification can inhibit apoptosis and promote dryness and tumorigenesis by affecting the lncRNA-miRNA-protein axis, which provides a potential target for the treatment of gastric cancer, but more samples are needed to verify and its role in other cancers can be further explored.
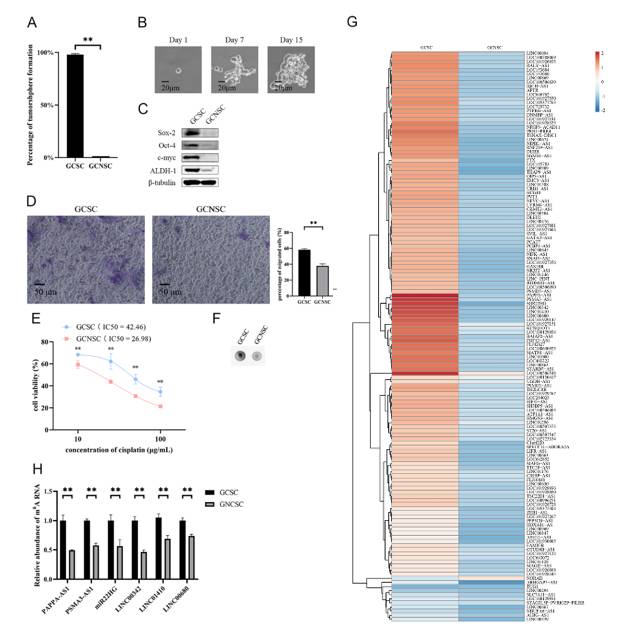 N6-methyladenosine lncRNAs in GCSCs (Ci et al., 2024)
N6-methyladenosine lncRNAs in GCSCs (Ci et al., 2024)
Reveal New Insights in Immunotherapy
One study focuses on the mechanism of methylation modification of RNA m6A in tumor development, and reveals that m6A modification mediated by METTL3 in myeloid cells affects tumor microenvironment by regulating macrophage polarization, thus promoting tumor growth and metastasis, and affecting the effect of immunotherapy, which provides new potential targets and theoretical basis for tumor treatment.
QRT-PCR, Western blot, ELISA, immunofluorescence staining, immunohistochemistry, chromatin immunoprecipitation (ChIP), m6A sequencing, RNA immunoprecipitation (RIP) and other techniques were used to detect gene expression, protein level, cytokine secretion, m6A modification level and protein-RNA interaction.
M6A sequencing found that deletion of Mettl3 affected m6A modification in BMDMs, and Spred2 was its target gene. The decrease of m6A modification reduced the translation of Spred2 mediated by YTHDF1, which in turn led to the up-regulation of ERK, NF-κB and STAT3 phosphorylation, and affected the function of macrophages.
METTL3 3 in macrophages can inhibit tumor, and its deletion can regulate macrophage polarization and related signal pathways by affecting m6A modification, reshape tumor microenvironment, promote tumor growth and metastasis, and weaken the effect of immunotherapy, suggesting that m6A regulation pathway of macrophages can be taken as the research direction of tumor treatment, but it is still necessary to further explore the related mechanism and the role of other cells in tumor microenvironment.
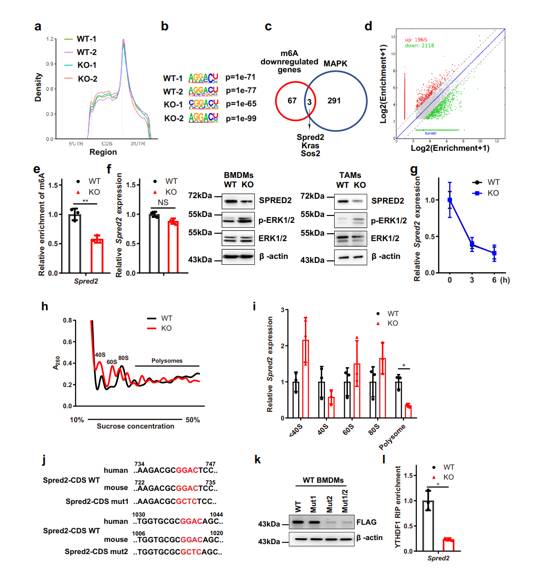 Metagene profiles of the m6A distribution across the transcriptome in WT and KO BMDMs (Yin et al., 2021)
Metagene profiles of the m6A distribution across the transcriptome in WT and KO BMDMs (Yin et al., 2021)
Explore Tumor Growth and Progress
One study focuses on the mechanism of RNA demethylase ALKBH5 in osteosarcoma, revealing the process of its influence on tumor growth and progress by regulating histone ubiquitination, which provides potential targets and theoretical basis for the treatment of osteosarcoma and other cancers.
MeRIP-seq analysis combined with RNA-seq data showed that USP22 was the key target gene of ALKBH5, and its MA level increased, its expression decreased and its mRNA stability decreased in silent cells of ALKBH5. Overexpression of ALKBH5 had the opposite result, and knock-down of METTL3 could up-regulate USP22 expression, which confirmed that ALKBH5 promoted its expression by maintaining the hypomethylation state of USP22.
ALKBH5 can regulate USP22 and RNF40 in osteosarcoma and other cancers, which is closely related to tumor growth and progress, and can be used as a potential therapeutic target for cancer. This study revealed for the first time the key role of MA modification in regulating histone ubiquitination and cancer occurrence and development, and clarified the regulation mechanism of ALKBH5-USP22/RNF40 axis, which provided a new idea for cancer treatment.
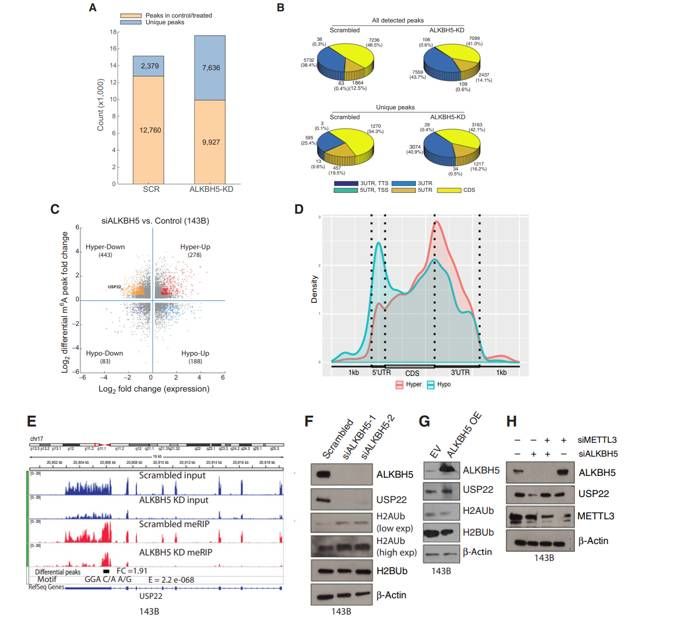 m6A methylation analyses of control and ALKBH5-silenced OS cells (Pooja et al., 2022)
m6A methylation analyses of control and ALKBH5-silenced OS cells (Pooja et al., 2022)
If you want to learn more about the MeRIP-seq, please refer to:
References
- Yang Wenlan, Zhao Yongliang and Yang Yungui. "Dynamic RNA methylation modifications and their regulatory role in mammalian development and disease." Science China Life Sciences (2024): 2084-2104. https://doi.org/10.1007/s11427-023-2526-2
- Ci Yuan, Zhang Yuan and Zhang Xiaobo. "Methylated lncRNAs suppress apoptosis of gastric cancer stem cells via the lncRNAs-miRNA protein axis." Cellular & Molecular Biology Letters (2024) 29: 51. https://doi.org/10.1186/s11658-024-00568-8
- Yin Huilong, Zhang Xiang, Yang Pengyuan, Zhang Xiaofeng and Zhang Rui. "RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming." Nature Communications (2021) 12:1394. https://doi.org/10.1038/s41467-021-21514-8
- Pooja Yadav, Panneerdoss Subbarayalu, Daisy Medina and Manjeet K Rao. "M6A RNA Methylation Regulates Histone Ubiquitination to Support Cancer Growth and Progression." Cancer Res (2022): 1872-1889. DOI: 10.1158/0008-5472.CAN-21-2106