
m1A RNA methylation is a newly discovered form of RNA methylation that refers to the methylation modification at the N1 position of adenosine in RNA molecules (N1-methyladenosine, m1A). Studies have shown that m1A is a post-transcriptional modification that is highly abundant in eukaryotic tRNA and rRNA, and recent research has also revealed its regulatory role in mRNA translation. As a novel type of RNA methylation, the functions and mechanisms of m1A modification are still being explored.
m1A-meRIP-seq Principle
Similar to the detection method for m6A RNA, MeRIP-seq technology is employed for m1A modification analysis. First, the m1A methylation level is quantified using LC-MS/MS. The principle of antibody enrichment is as follows: the m1A-specific antibody is co-incubated with randomly fragmented RNA fragments, and the methylated fragments are captured for sequencing. Simultaneously, a control sample (Input) is sequenced, which consists of RNA fragments without undergoing the immunoprecipitation (IP) reaction. The control sample is used to eliminate the background of non-specifically captured methylated fragments. Additionally, the characteristic of m1A causing mismatched base pairs is utilized. The captured fragments from antibody enrichment are subjected to demethylation using the AlkB demethylase. By comparing the results of (-) demethylase and (+) demethylase treatments, the signals of base mutations are captured to achieve single-nucleotide resolution identification of m1A methylation sites. By comparing the sequence fragments between the immunoprecipitated IP sample and the Input sample, the m1A RNA methylation modification sites are mapped onto the transcriptome. With the aid of RNA-seq data, not only can the degree of m1A methylation in the sample be calculated, but also the specific site information of m1A methylation modification can be determined.
CD Genomics provides m1A-seq technologies suitable for different research needs:
-m1A Methylation
- Bulk mRNA Methylation Sequencing (m1A-seq) m1A Methylation
- Bulk mRNA + lncRNA Methylation Sequencing (lnc-m1A-seq) m1A Methylation
- Trace amount of mRNA + lncRNA Methylation Sequencing (Micro-lnc-m1A-seq)
Technical Advantages
Single-nucleotide resolution detection of m1A methylation modification.
High specificity: m1A RNA-seq utilizes commercial m1A antibodies for specific enrichment of m1A-modified fragments.
One-stop service: Customers only need to provide cells, tissues, body fluids, or total RNA, and CD Genomics will complete the entire service process from m1A RNA-seq enrichment, library preparation, sequencing, to data analysis.
Professional bioinformatics analysis: CD Genomics has a powerful bioinformatics team that can meet various in-depth data analysis requirements from customers.
m1A-seq Analysis Workflow
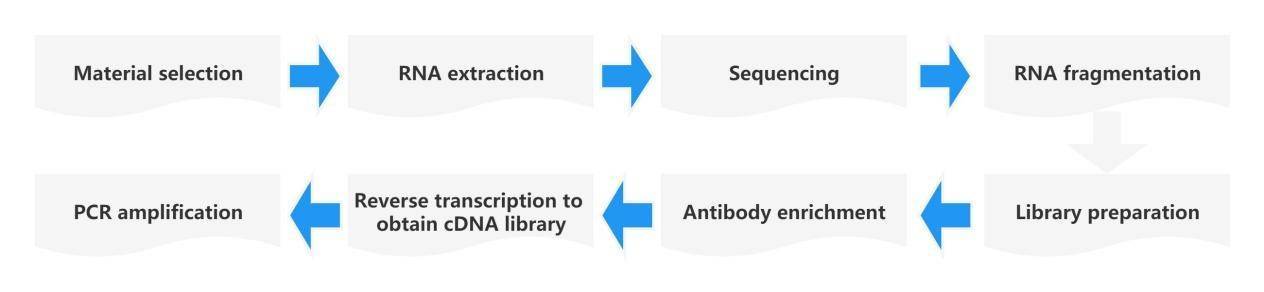
m1A-seq Analysis Content
| Analysis Content | |
|---|---|
| Standard Analysis | Quality control and assessment: Data quality control Alignment statistics Insert fragment length statistics Whole-genome coverage statistics Sample saturation curve |
| Identification and statistics of m1A enrichment regions (peaks) | Identification and annotation of m1A enrichment regions (peaks) Basic statistics of m1A enrichment regions Distribution of m1A enrichment regions on chromosomes Distribution of m1A enrichment regions on genomic elements Functional enrichment analysis of genes associated with m1A enrichment regions Density of m1A enrichment regions on genomic element Motif identification in m1A modification regions 8. Specific gene peak visualization |
| Identification and statistics of differentially modified m1A | Analysis of peak overlap within the group Analysis of overlap between peaks and genes within the group Identification and annotation of differentially modified m1A Basic statistics of differentially modified m1A Distribution of differentially modified m1A on chromosomes Distribution of differentially modified m1A on genomic element Enrichment analysis of genes associated with differentially modified m1A Density of differentially modified m1A on genomic elements Motif identification in differentially modified m1A regions |
| Expression level analysis | Identification of lncRNAs Identification of circRNAs Analysis of gene expression levels Analysis of gene expression level density Clustering analysis based on expression profiles Principal component analysis based on expression profiles Correlation analysis based on expression profiles |
| Identification and enrichment analysis of differentially expressed genes | Identification of differentially expressed genes Functional enrichment analysis of differentially expressed genes |
| Analysis of differential m1A modifications and associated genes | Analysis of the association between differential m1A modifications and differential genes |
Sample Requirements
| Sample Type | Depleted and DNase-treated total RNA samples |
|---|---|
| Sample Amount | ≥10μg |
| Sample Concentration | Recommended ≥250 ng/μL |
| Sample Integrity | RIN ≥7, Ratio 28S/18S ≥0.7 |
| Additional Requirements | For certain sample sources (such as fluid samples, insect samples, aquatic organisms, etc.), there are no specific requirements for RIN and Ratio 28S/18S. |
Case 1: The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA
Journal: Nature
Impact Factor: 41.456
Background
m1A RNA methylation occurs in the Watson-Crick region and may affect RNA base pairing. It also promotes adenosine modification with a positive charge under physiological conditions, which can significantly alter RNA structure and protein-RNA interactions.
Methods
The researchers performed antibody-based enrichment of RNA methylation immunoprecipitation sequencing (MeRIP-seq, m1A-seq) on human HeLa (cervical adenocarcinoma), HepG2 (liver hepatocellular carcinoma), and HEK293 (embryonic kidney) cell lines (ATCC), as well as primary mouse embryonic fibroblast (MEF) cells (C57BL/6, ATCC). They localized m1A sites in the transcriptome and coupled them with Dimroth rearrangement to obtain a high-resolution m1A map.
Results
The m1A-seq results showed enrichment of m1A around the translation initiation site upstream of the first splice junction: m1A tended to modify more structured regions around the translation initiation site, which can dynamically respond to physiological conditions and are positively correlated with protein generation. These specific features are highly conserved in both mouse and human cells, demonstrating the role of m1A in facilitating mRNA methylation translation.
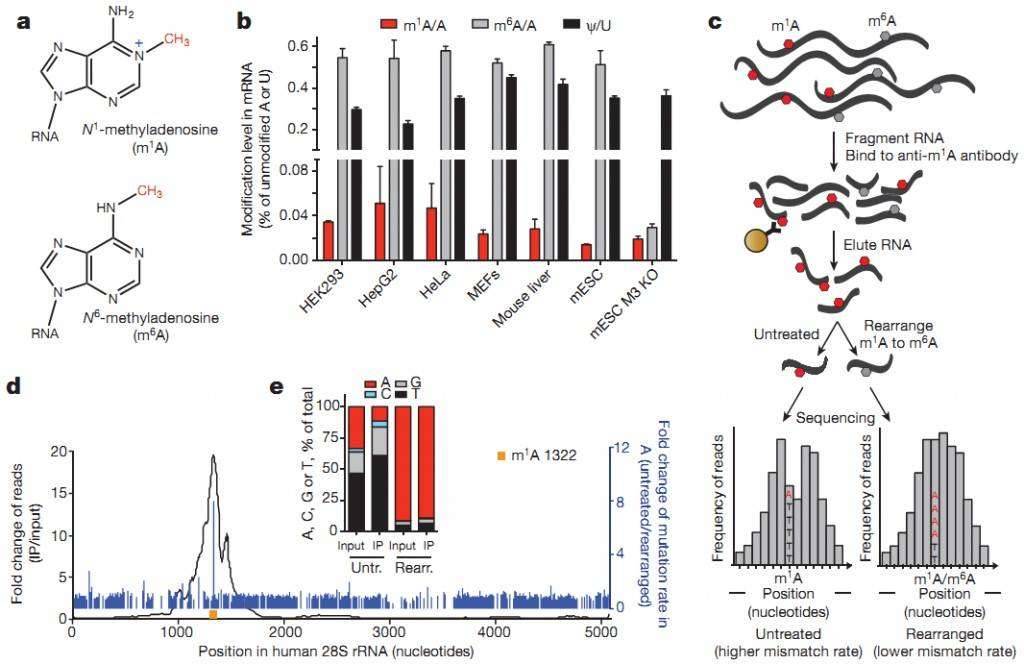 Figure 1: m1A-seq reveals the association of m1A with translation initiation sites (TIS) in the human transcriptome.
Figure 1: m1A-seq reveals the association of m1A with translation initiation sites (TIS) in the human transcriptome.
Case 2: Title: Base-Resolution Mapping Reveals Distinct m1A Methylome in Nuclear- and Mitochondrial-Encoded Transcripts
Journal: Molecular Cell
Impact Factor: 15.59
This study employed a base-resolution method called "m1A-MAP" to identify m1A methylated sites in nuclear and mitochondrial transcripts. The results revealed that the majority of m1A methylation modifications are enriched in the 5' untranslated region (UTR) of mRNA, while a small subset of m1A sites conforming to the "GUUCRA" motif are modified by a known methyltransferase complex, TRMT6/61A. Furthermore, significant m1A methylation modifications were observed in mitochondrial-encoded transcripts. The study also demonstrated that m1A located in the 5' UTR, particularly at the first and second positions of the transcripts, can enhance mRNA translation, while m1A in the coding region can inhibit translation. Therefore, m1A sequencing not only revealed distinct types of m1A methylation modifications in nuclear and mitochondrial-encoded transcripts but also provided an important tool for further functional validation of m1A methylation.
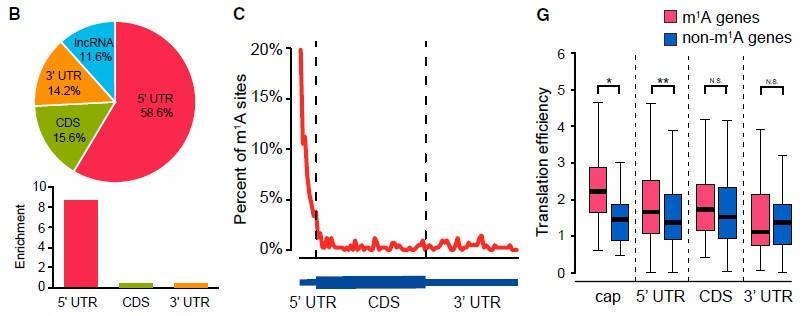 Figure 2: Distribution of m1A RNA modifications in the human transcriptome and their impact on translation efficiency.
Figure 2: Distribution of m1A RNA modifications in the human transcriptome and their impact on translation efficiency.
Reference
- Dominissini D, et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016 Feb 25;530(7591):441-6. pii: nature16998.