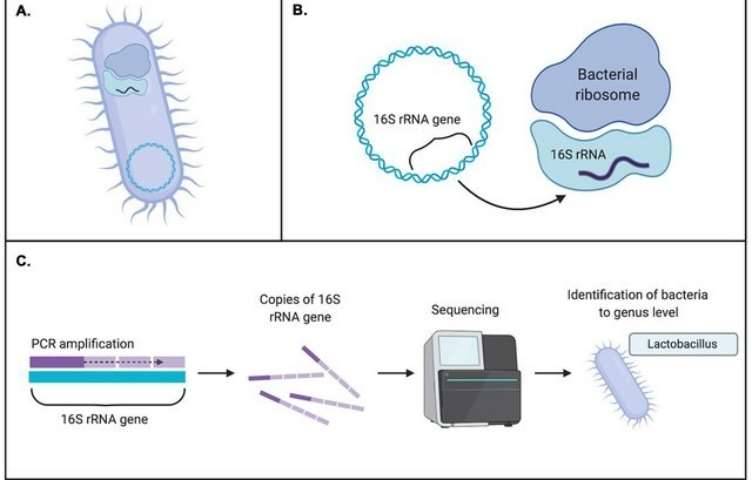
Microbial entities, by their minuscule nature, necessitate observation through the aid of scientific instruments. Although individually imperceptible to the naked eye, their collective abundance is vast, extending widely across the surrounding environment as well as within the bodies of flora and fauna. Remarkably, these microorganisms wield a substantial influence on human health, animal development, and the cultivation of plants. Their pervasive presence underscores their pivotal role in the intricate tapestry of biological interactions.
How to Investigate Microorganisms?
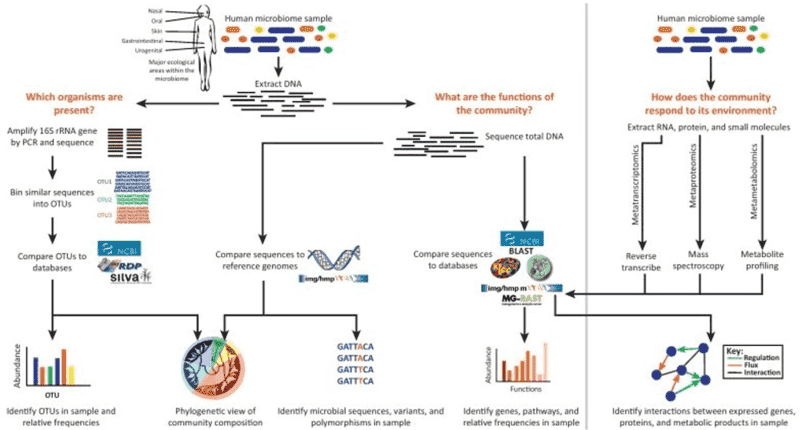
The study of microbial communities encompasses three pivotal dimensions: microorganisms themselves, along with their DNA and mRNA constituents. The in-depth exploration of microbial communities relies upon a diverse array of technological methodologies. Among these, amplicon sequencing, metagenomic sequencing, and metatranscriptomic sequencing play central roles in elucidating microbial genetic and transcriptional landscapes. This article will intricately expound upon these three techniques, delineating their applications in the realm of microbial community research.
I. Amplicon Sequencing
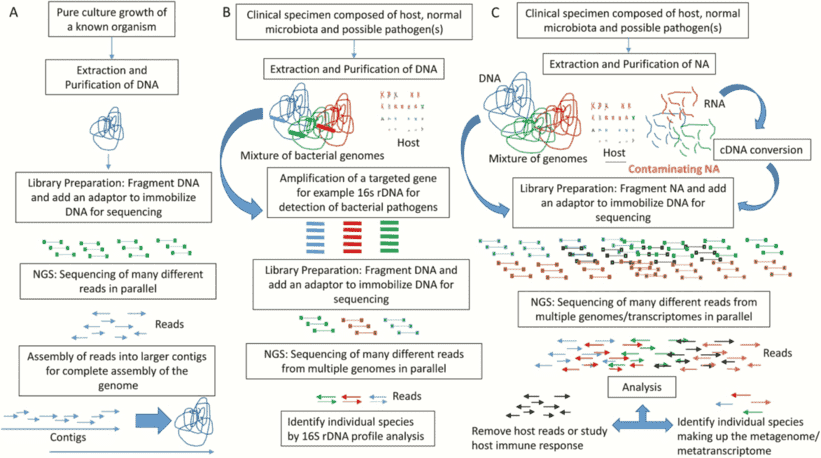
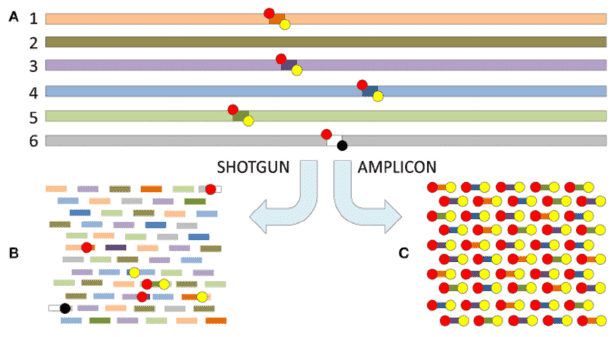
Amplicon sequencing entails the amplification of specific DNA fragments through PCR technology, followed by the sequencing of these amplified products. This methodology is primarily employed to address the question of "which microorganisms are present." By amplifying and sequencing high-variable regions of 16S, 18S, ITS, and functional genes within a sample, microbial composition can be identified at the genus/species level. It is imperative to note that the target of amplicon sequencing is the genetic material in the form of DNA.
| Product | Research Targets | Next-generation sequencing regions | Long read sequencing region |
|---|---|---|---|
| 16S sequencing | Bacteria/Archaea | V3-V4 region | Full length |
| 18S sequencing | Eukaryotic microorganisms | V4 region | |
| ITS sequencing | Fungi | ITS1 region | |
| Functional gene amplicon sequencing | Other target genes | Target genes |
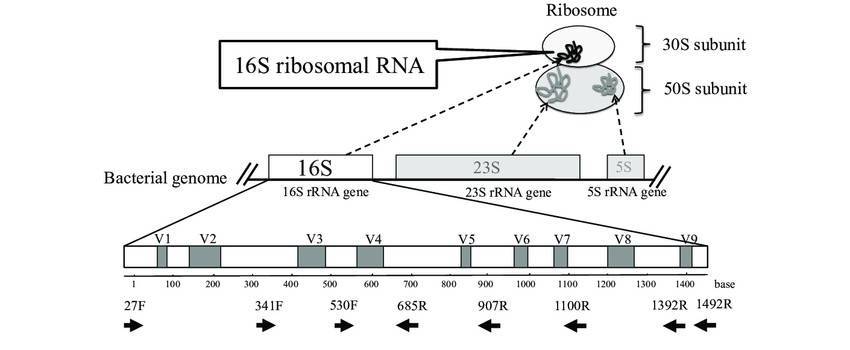
The ribosomes of prokaryotes consist of 5S rRNA, 23S rRNA, and 16S rRNA. Among these, the gene encoding 16S rRNA possesses distinct characteristics: ubiquity in prokaryotes, moderate size (1540 nt), high copy numbers, ease of template acquisition, and inclusion of both highly conserved and variable regions. These features render the 16S rRNA gene particularly well-suited for bacterial diversity analysis.
The sequence of 16S rDNA is characterized by the presence of nine conserved regions juxtaposed with nine variable regions. The preference for a specific sequencing region is largely influenced by the inherent attributes of the sample under investigation.
More precisely, for bacterial diversity assessment, the V3-V4 region of the 16S rDNA sequence is frequently utilized. This particular region is particularly effective when analyzing intestinal specimens due to its high level of suitability.
In contrast, the V5-V7 region proves beneficial when examining samples likely to contain plant host contaminants, specifically, endophytic bacteria, due to its unique attributes.
The V4-V5 region, however, is distinctly suitable when dealing with environmental specimens such as soil and sediment. This suitability arises from the region's inherent characteristics that enable elucidation of environmental microbial diversity present in these samples.
Therefore, these regions are selected based on their unique attributes that allow for effective bacterial diversity assessment, thereby contributing to the ever-expanding field of microbiome research. The process of choosing an appropriate sequencing region is rooted in its applicability and potential to deliver accurate, insightful data instrumental in advancing our understanding of bacterial diversity.
16S Sequencing Workflow
The sequencing workflow of amplicon sequencing technologies generally exhibits fundamental consistency. Taking the 16S amplicon sequencing process as an illustrative example, we can delineate its procedural steps in detail. Initially, total DNA is extracted from the sample. Subsequently, primers designed based on conserved regions are employed for PCR amplification. Following this, the amplified products undergo purification, quantification, and normalization, culminating in the formation of a sequencing library. Subsequent steps involve quality control, sequencing, and data analysis. The 16S amplicon sequencing workflow is illustrated in the figure below.

Within eukaryotes, the V4 region database of 18S rDNA encapsulates the most comprehensive information and demonstrates excellent classification efficacy. Consequently, it is widely acknowledged as the optimal choice for the annotation and analysis of the 18S rRNA gene, particularly for taxa such as algae and protozoa within the eukaryotic domain.
The Internal Transcribed Spacer (ITS) sequence, situated between the fungal 18S, 5.8S, and 28S rRNA genes, comprises two distinct segments: ITS1 (350 bp) and ITS2 (400 bp).ITS1 is conventionally favored, whereas ITS2 finds applicability in the identification of fungi within samples such as those derived from fermentation processes and environmental matrices like soil.
Functional Gene Amplicon Sequencing
Functional microorganisms refer to microbes endowed with specific biological functions. While these microorganisms may exhibit considerable taxonomic diversity, they share analogous genes that enable them to perform similar functions. Consequently, genes that facilitate the expression of specific functions in functional bacteria, such as nxrA, nirS/nirK, amoA, dsrB, nifH, and others, are collectively referred to as functional genes. The process of primer design, amplification, and sequencing targeting these functional genes is termed Functional Gene Amplicon Sequencing.
| Functional Microorganisms | Functional Genes |
|---|---|
| Nitrogen-fixing microorganisms | nifH |
| Carbon-fixing microorganisms | cbbL and cbbM |
| Nitrite-oxidizing bacteria (NOB) | nxrA |
| Ammonia-oxidizing bacteria (AOB) | amoA |
| Sulfate-reducing bacteria (SRB) | dsrB |
| Methane-oxidizing bacteria | pmoA |
| ... | ... |
II. Metagenomics
In 1998, Jo Handelsman introduced the concept of metagenomics, representing the collective genomic content of all microorganisms in an environment. Diverging from amplicon sequencing, metagenomic sequencing involves the sequencing of the genomes of all microbial populations within a sample, rather than singularly focusing on specific populations. This approach is principally employed to address the question of "what microbial communities are capable of."
Through metagenomic sequencing, we can delve into the intricate exploration of microbial diversity, population structures, evolutionary relationships, functional activities, inter-species interactions, and their interplay with the environment. This method furnishes a more comprehensive understanding of both the structural composition and functional dynamics of microbial communities. Its application significantly contributes to a profound comprehension of the roles and functionalities of microorganisms in the natural world.
Moreover, metagenomic sequencing extends its utility beyond the realm of microbial ecology, finding relevance in diverse fields such as environmental monitoring, disease diagnosis and treatment, as well as the development of bioenergy resources. Hence, metagenomic research holds substantial and impactful applications across multiple domains.
Metagenomic Sequencing Workflow
The metagenomic sequencing workflow exhibits distinct differences from amplicon sequencing procedures. In metagenomic sequencing, the necessity for PCR amplification is obviated. Instead, DNA extraction directly from the sample is conducted, followed by subsequent processes such as library construction and sequencing. This streamlined approach proves to be comparatively more concise and efficient, providing a more accurate representation of the authentic microbial community within the sample. This methodology underscores a commitment to capturing the genuine intricacies of microbial populations without the intermediary influence of PCR amplification steps.

III. Metatranscriptomics
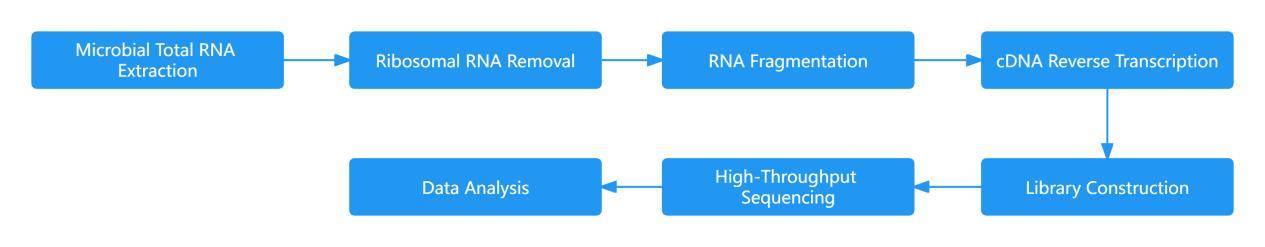
Metatranscriptomics aims to address the question of "what microbes are actively doing." Under specific conditions, this approach involves high-throughput sequencing of all microbial RNA present in environmental or tissue samples, providing direct access to the transcriptomic information of the entire microbial community within the sample. By studying the transcriptional changes in complex microbial communities at the RNA level, this method unveils the potential for discovering novel genes.
Metatranscriptomic Sequencing Workflow
The central objective of metatranscriptomic research is to capture the collective RNA repertoire of all microbes within a given sample, with mRNA enrichment standing as a pivotal step in this process. During the enrichment phase, methods typically involve the removal of rRNA to ensure the acquisition of high-quality mRNA samples. Once mRNA enrichment is achieved, subsequent steps in constructing the sequencing library align with standard transcriptomic sequencing procedures.
This meticulously designed workflow not only upholds the rigor and scientific precision of metatranscriptomic sequencing but also lends robust support to the in-depth exploration of transcriptional activities within microbial communities. The incorporation of rRNA removal strategies ensures the fidelity of the metatranscriptomic data, thereby facilitating a comprehensive investigation into the intricate transcriptional dynamics of microbial populations.
IV. Comparative Analysis of Technical Methods
In the selection of experimental approaches, a comprehensive assessment considering the characteristics of the samples and the research objectives is imperative for these three distinct technical methods. The ensuing analysis provides a comparative evaluation of amplicon sequencing, metatranscriptomics, and metagenomics:
| Amplicon Sequencing | Metagenomics | Metatranscriptomics | |
|---|---|---|---|
| PCR Amplification | Yes | No | No |
| Advantages | - No host contamination issues - Cost-effective, suitable for large-scale studies |
- No amplification required, includes all DNA | - Provides information on gene expression differences |
| Cost | Low | High | High |
| Species Identification | Next-gen (genus) or long read (species) | Species | - |
| Disadvantages | - PCR amplification has preferences - Limited functional information - Primers and variable regions affect results - Cannot distinguish living vs. non-living DNA |
- Prone to host contamination - High sequencing throughput - Cannot distinguish living vs. non-living DNA - Longer genome assembly cycle |
- Prone to host contamination - Risk of ribosomal RNA contamination - Requires integration with DNA sequencing data for bacterial abundance and transcription rates |
V. How to Choose?
In light of diverse experimental objectives, we proffer the following methodological recommendations:
| Method | Description |
|---|---|
| Amplicon Sequencing Technique | Recommended for investigating microbial diversity and community composition within the sample, without necessitating a deeper level of functional analysis. |
| Metagenomic Sequencing Technique | Advisable when the research demands an exploration beyond microbial species, delving into the study of related genes and functionalities. |
| Integrated Analysis of Amplicon and Metagenomic Sequencing | Recognizes divergent research goals and depths between amplicon and metagenomic sequencing. A judicious approach involves initial screening using amplicon sequencing for multiple replicates or large sample sizes, eliminating outliers. Subsequently, samples with high reproducibility undergo metagenomic sequencing for more in-depth exploration. |
| Integrated Analysis of Amplicon, Metagenomic, and Metatranscriptomic Sequencing | Leverages the synergy of multi-omics analysis for a comprehensive understanding of microbial communities within the sample. This approach addresses questions related to the identification of microbial entities, their capabilities, and ongoing activities, culminating in a holistic investigative framework. |
VII. Application
(1) In the realm of medicine, our focus is on the study of human microbiota, including vaginal and skin microbial communities. Simultaneously, we delve into in-depth research on diseases, encompassing disease diagnosis and prediction, as well as aspects related to metabolic disorders, digestive diseases, autoimmune diseases, and resistance gene detection. Furthermore, our dedication extends to cancer research, involving the diagnosis and prediction of cancer types, as well as the discovery of therapeutic targets.
(2) In the field of environmental studies, our emphasis lies in the investigation of microorganisms within specific environments. We are dedicated to addressing relevant environmental challenges, including the fermentation processing of organic fertilizers, sewage treatment, petroleum degradation, and issues pertaining to aquatic and marine environments.
(3) In the agricultural domain, our focus revolves around the symbiotic relationships between microorganisms in the intestines, rumen, and various facets of animal reproduction, growth, development, nutritional health, immune response, and disease treatment. Simultaneously, we investigate the interactions between rhizospheric microorganisms and plants, as well as issues related to agricultural cultivation/fertilization practices and soil microbiota.
(4) In the realm of bioenergy research, our dedication is directed towards the study of specialized bacterial strains, gene exploration, and the development of engineered microbes.
(5) In the domain of extreme environments, our focus is dedicated to the study of microorganisms thriving in extreme conditions, such as archaea.
VIII. Case Studies
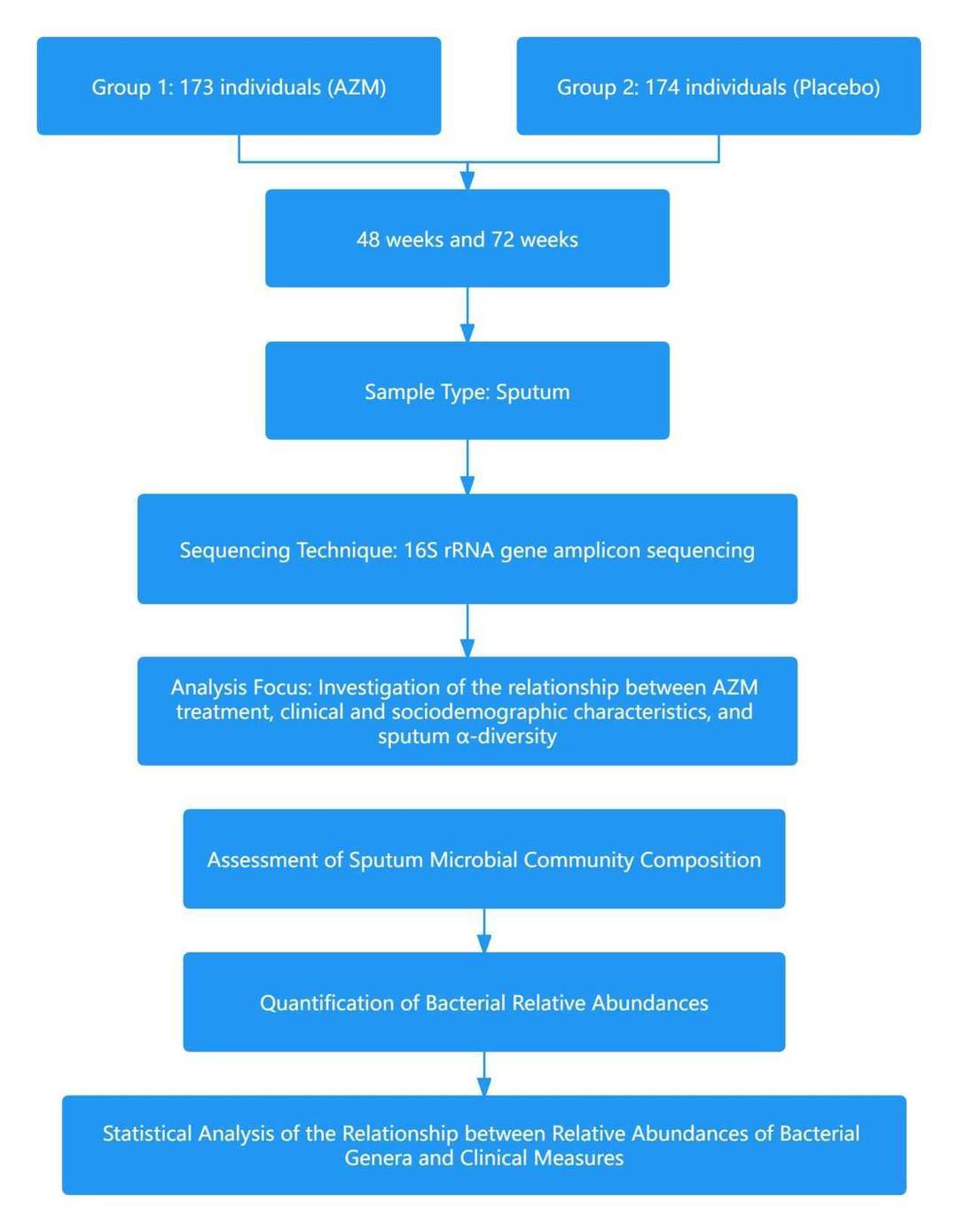
Case 1: Sputum bacterial load and bacterial composition correlate with lung function and are altered by long term azithromycin treatment in children with HIV associated chronic lung disease
Chronic lung disease directly associated with HIV (HCLD) represents the most widespread chronic complication directly related to HIV, accounting for over half of all HIV-related mortality. Manifestations of this condition include dyspnea, diminished exercise tolerance, persistent fatigue, and chronic coughing. The mechanisms underlying this HIV-related obstructive bronchiolitis remain a subject of ongoing investigation. Nevertheless, recent experimental findings have proposed the potential of extended Azithromycin (AZM) treatment as a method to reduce the incidence of acute respiratory exacerbations among pediatric and adolescent populations suffering from HIV-associated chronic lung disease (HCLD). The implications of this treatment strategy on the bacterial flora of the respiratory system, however, is an area requiring further elucidation.
In February 2023, a research article headed by Mark P. Nicol of the University of Cape Town in South Africa, was published in the esteemed journal "Microbiome". The publication is titled "Sputum bacterial load and bacterial composition correlate with lung function and are altered by long-term azithromycin treatment in children with HIV-associated chronic lung disease." The findings of the research signify that prolonged AZM therapy aids in retaining the bacterial diversity in sputum and lessens the relative prevalence of HCLD-associated bacteria such as Haemophilus and Moraxella. These microbiological modifications are linked with enhanced pulmonary function, which potentially elucidates the observed reduction in respiratory exacerbations in children suffering from HCLD post AZM therapy.
Technical Approach:
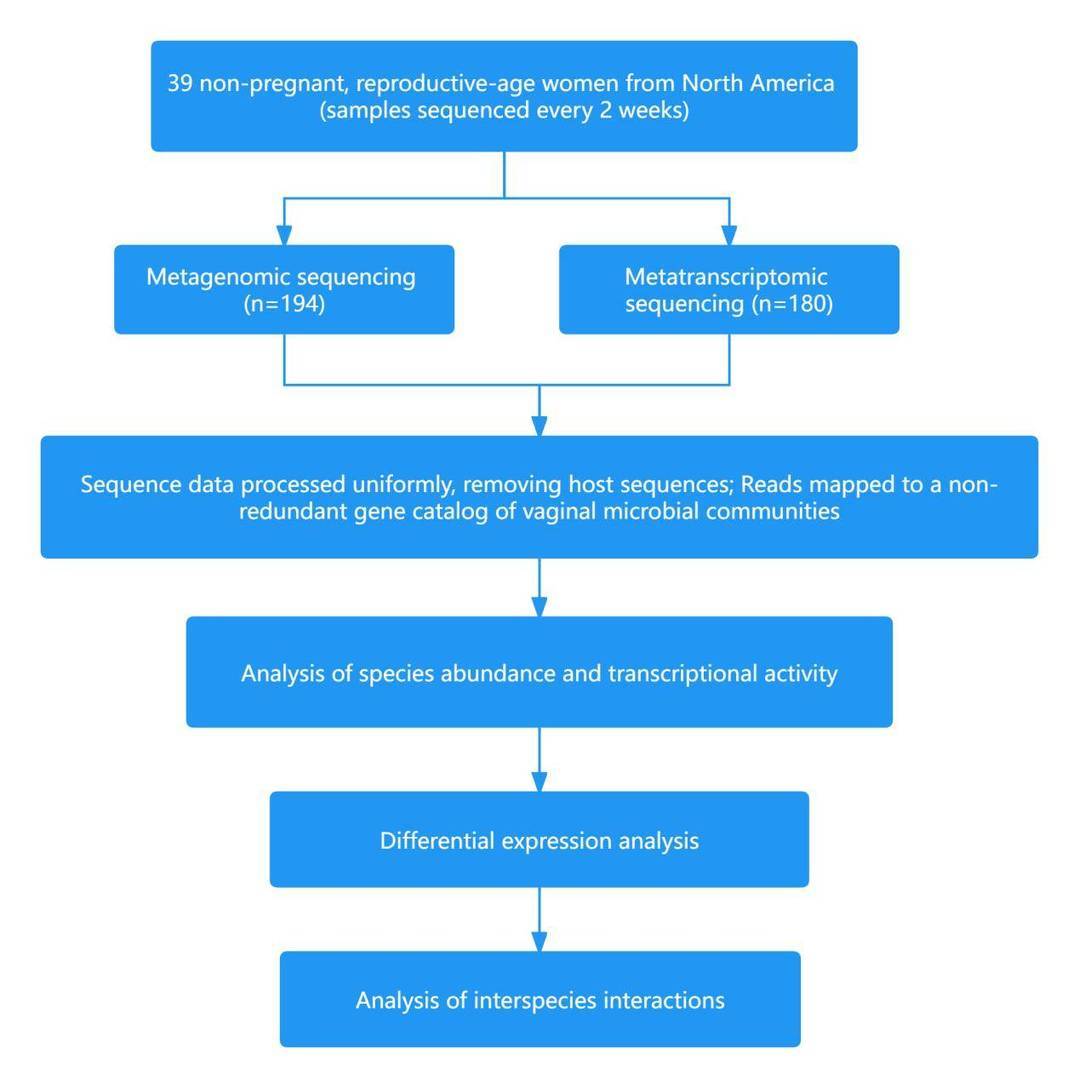
Case 2: Insight into the ecology of vaginal bacteria through integrative analyses of metagenomic and metatranscriptomic data
The microbial community within the human body stands as a critical determinant of health. In women of childbearing age, the vaginal microbial community can be broadly categorized into five distinct Community State Types (CSTs). Among these, four are dominated by lactic acid bacteria (CST I - Lactobacillus crispatus, CST II - Lactobacillus gasseri, CST III - Lactobacillus iners, CST V - Lactobacillus jensenii), with high relative abundances. CST IV primarily consists of a diverse mix of microbes, including strict anaerobes and facultative anaerobes. Vaginal bacterial communities predominantly composed of lactic acid bacteria have been associated with a reduced risk of various adverse health outcomes, although the factors driving these changes remain unclear.
In March 2022, the team led by Jacques Ravel at the University of Maryland School of Medicine published a paper in the Genome Biology journal titled "Insight into the ecology of vaginal bacteria through integrative analyses of metagenomic and metatranscriptomic data." The study revealed that vaginal transcriptomic data could predict future changes in community composition, aiding in the early diagnosis of vaginal diseases such as symptomatic bacterial vaginosis. Furthermore, the results from metatranscriptomic data guide the development of innovative strategies to modulate the composition and activities of vaginal microbial communities, aiming to restore and maintain an optimal protective environment.
Technical Approach:
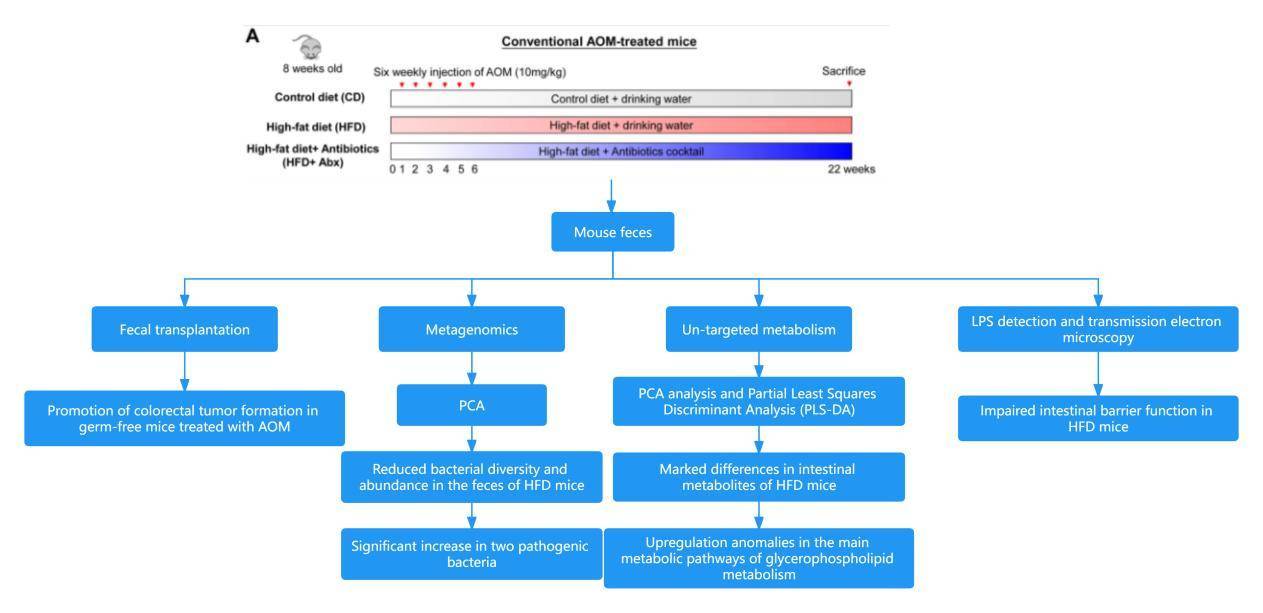
Case 3: High-Fat Diet Promotes Colorectal Tumorigenesis Through Modulating Gut Microbiota and Metabolites
Colorectal cancer (CRC) stands as the third most prevalent and lethal cancer worldwide, and excessive dietary fat intake is intricately linked to an increased risk of CRC. Unlike other types of cancer, colorectal cancer directly interacts with trillions of gut microbiota during the tumor development process. The composition of the gut microbiota is influenced by various factors, including diet, medications, and genetic alterations. Alterations in the microbial spectrum can lead to ecological imbalance and contribute to CRC. However, the underlying mechanisms between dietary fat intake and the development of colorectal cancer remain unclear.
The paper "High-Fat Diet Promotes Colorectal Tumorigenesis through Modulating Gut Microbiota and Metabolites" penned by Professor Yu Jun, flanked by his scholarly team from The Chinese University of Hong Kong, was showcased in the reputable Gastroenterology journal in January 2022. The research establishes the link between indulgence in a high-fat diet (HFD) and the heightened probability of succumbing to colorectal cancer. The study unveils how an HFD can incite an ecological imbalance within the intestinal microbiota, disrupt metabolic harmony, and compromise the integrity of the intestinal epithelial barrier in mice. Paradoxically, these aberrations brought upon by an HFD can trigger colorectal carcinogenesis by amplifying the expression of oncogenes while undermining the intestinal barrier's performance. Consequently, the study brings forth the idea of manipulating gut microbiota and metabolic patterns as potential therapeutic stratagems, particularly for forestalling and mitigating HFD-induced colorectal cancer.
Technical Approach:
Conclusion
The advent of methodologies like amplicon sequencing, metagenomics, and metatranscriptomics has revolutionized the examination of microbial diversity and functionalities across varied environmental samples. Enabling a more nuanced understanding of diseases, fostering insights into the phenology of flora and fauna, and contributing significantly towards environmental conservation initiatives, these techniques exemplify human ingenuity in scientific research advancement.
References
- Liu YX,Qin Y,Chen T, et al. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell. 2021;12 (5):315-330. doi:10.1007/s13238-020-00724-8
- Abotsi RE,Dube FS,Rehman AM, et al. Sputum bacterial load and bacterial composition correlate with lung function and are altered by long-term azithromycin treatment in children with HIV-associated chronic lung disease. Microbiome. 2023;11 (1):29. doi:10.1186/s40168-023-01460-x
- France MT,Fu L,Rutt L, et al. Insight into the ecology of vaginal bacteria through integrative analyses of metagenomic and metatranscriptomic data. Genome Biol. 2022;23 (1):66. doi:10.1186/s13059-022-02635-9
- Zhao W,Chen Z,Yang X, et al. Metagenomics reveal arbuscular mycorrhizal fungi altering functional gene expression of rhizosphere microbial community to enhance Iris tectorum's resistance to Cr stress. Sci Total Environ. 2023;895:164970. doi:10.1016/j.scitotenv.2023.164970
- Liu Y,Wang H,Qian X, et al. Metagenomics insights into responses of rhizobacteria and their alleviation role in licorice allelopathy. Microbiome. 2023;11 (1):109. doi:10.1186/s40168-023-01511-3
- Yang J,Wei H,Zhou Y, et al. High-Fat Diet Promotes Colorectal Tumorigenesis Through Modulating Gut Microbiota and Metabolites. Gastroenterology. 2022;162 (1):135-149.e2. doi:10.1053/j.gastro.2021.08.041