Introduction to Full-Length 16S/18S/ITS Sequencing
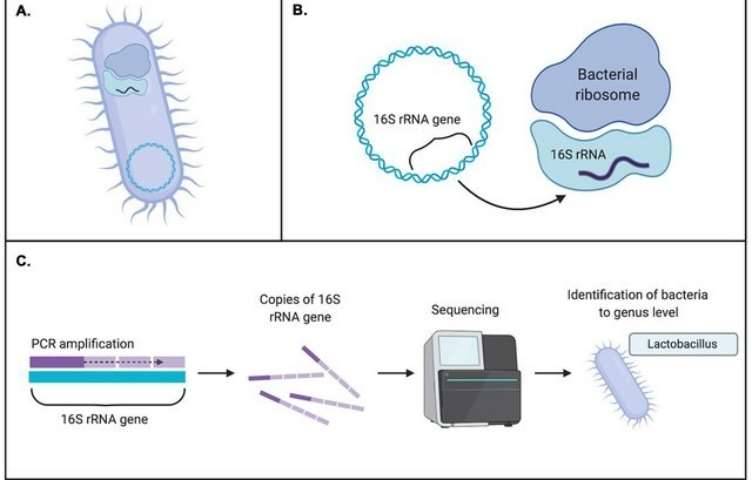
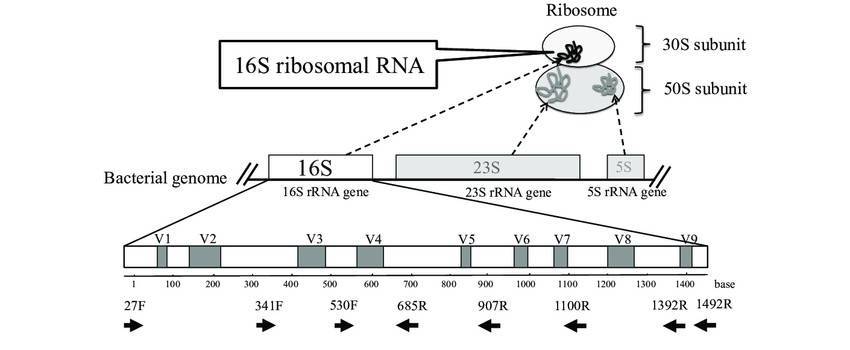
The prevailing methods for investigating microbial diversity predominantly hinge on the capabilities of second-generation sequencing (SGS). Among these, short-read strategies like paired-end sequencing (e.g., PE250) dominate. These approaches zero in on hypervariable regions of bacterial or fungal genetic material—think of the V3 and V4 segments of the 16S ribosomal RNA (rRNA) gene, the V4 region of 18S rRNA, or even the ITS1 (internal transcribed spacer 1). Through these targeted sequences, researchers untangle the taxonomic tapestry of microbial communities and infer their functional genomic blueprints from environmental samples.
A Leap Forward: Long-Read Sequencing (LRS)
Enter long-read sequencing (LRS)—a game-changer that has not only earned accolades like Nature Methods' "Method of the Year" but also transformed how microbial diversity is studied. By embracing technologies such as the PacBio platform and SMRT (single-molecule real-time) sequencing, LRS delivers read lengths stretching up to 20 kilobases (kb) and HiFi accuracies surpassing 99.9%. This leap in precision allows scientists to peel back the intricate layers of microbial community structures, capturing nuances previously blurred by shorter reads.
Short Reads vs. Long Reads: A Tug of Precision
SGS methods, while highly effective for genus-level insights, often fall short when a more granular view is needed. Here's where long-read sequencing steals the spotlight: full-length microbial diversity sequencing captures the entirety of key genetic markers, such as the complete 16S rRNA gene. This holistic view enables not just species-level classification but also the detection of fine-scale variations and tracking of specific taxa (Xia et al., 2019). In essence, it transforms the sequencing landscape from a sketch into a masterpiece.
Rewriting Research Potential
The significance of full-length sequencing extends far beyond ecology—it ripples into environmental science and even clinical applications. By unraveling microbial networks with unmatched clarity, it's paving the way for breakthroughs in understanding ecosystem dynamics and microbial roles in health and disease. The era of piecing together fragmented genetic data is giving way to an age of seamless insight.
Whether you're mapping microbial inhabitants of a pristine glacier or dissecting the neonatal gut microbiome, full-length sequencing is not just a method—it's a revolution in how we view microbial diversity.
Species-Level Specificity in Microbial Diversity Analysis
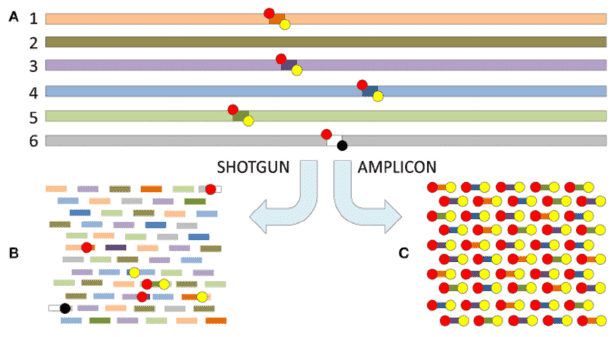
As demonstrated in a seminal 2019 study published in Nature Communications, full-length 16S ribosomal RNA (rRNA) sequencing offers superior taxonomic resolution compared to short-read sequencing methods. Researchers have systematically evaluated and compared short-read and long-read sequencing approaches, summarized in Table 1. Second-generation sequencing (SGS) methods rely on partial variations within conserved regions and primarily support analyses at the genus level or higher. In contrast, third-generation sequencing (TGS) captures the full variability of conserved regions, enabling species-level taxonomic annotation.
Moreover, third-generation full-length amplicon sequencing facilitates the detection and tracking of specific species within environmental samples, as well as the confirmation of previously undetected strains. Representative results are illustrated in Figure 1.
Table 1: Summary of Sequencing Methods for Microbial Diversity Analysis
| Method |
Sequencing Type |
Sequencing Platform |
Read Lengths (bp) |
Reads per Sample (M) |
Output |
Data Processing Required |
Cost per Sample |
| SRA (V3-V4) |
Amplicon |
Illumina MiSeq |
250 or 300 |
0.03–0.2 |
Taxonomic profiling (genus level) |
Low |
$ |
| LRA (16S-ITS-23S) |
Amplicon |
PacBio Sequel II |
~2500 |
0.01–0.03 |
Taxonomic profiling (strain or species level) |
Low |
$ |
Case Studies: Neonatal Gut Microbiome Analysis
A recent investigation amplified and sequenced stool samples from neonates, comparing the results against microbial reference databases. The study successfully identified the species-level bacterial composition of the neonatal gut. Unique strains with distinct amplicons were revealed, producing specific amplicon sequence variant (ASV) fingerprinting profiles.
Further analysis included longitudinal time series data from the stool microbiota of individual infants and twins. These data elucidated the bacterial community structure in preterm infants, characterized by simplified microbiota, thereby offering insights into the dynamics of gut microbial composition in early life.
Discussion
The results underscore the species-level specificity afforded by third-generation full-length sequencing methods. Compared to SGS, TGS provides a more comprehensive representation of microbial diversity, facilitating advanced ecological and clinical research.
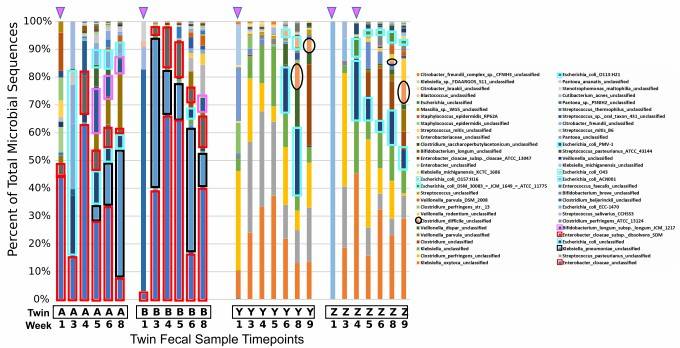 Figure 1 Overview of bacterial communities
Figure 1 Overview of bacterial communities
Strain-Level Single Nucleotide Specificity in Microbial Diversity Studies
A study published in Nucleic Acids Research explored the construction of an artificial bacterial community comprising eight phylogenetically distant bacterial species. By analyzing the resulting data, the researchers identified 29 amplicon sequence variants (ASVs), each annotated to its corresponding genus and species. The results, as shown in Figure 2, underscore the precision of full-length 16S ribosomal RNA (rRNA) gene sequencing in capturing ASV diversity.
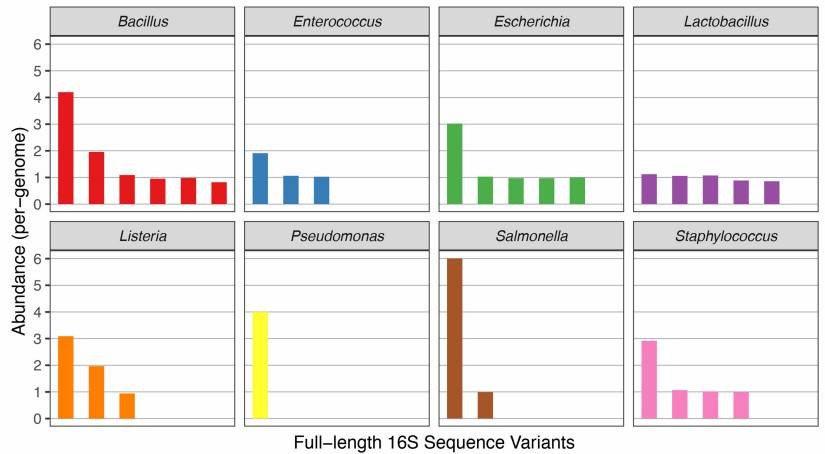 Figure 2: Abundance of Amplicon Sequence Variants (ASVs) Detected in a Simulated Community
Figure 2: Abundance of Amplicon Sequence Variants (ASVs) Detected in a Simulated Community
Distinguishing Closely Related Strains
In addition to distinguishing phylogenetically distant species, third-generation sequencing technologies have demonstrated the capability to differentiate closely related bacterial strains. For instance, as shown in Figure 3, researchers tracked the distribution of Klebsiella strains within neonatal stool samples. The study identified 54 distinct ASVs corresponding to Klebsiella and analyzed their abundance across various samples. The results indicated that samples containing the same Klebsiella strain exhibited similar ASV distribution patterns, reinforcing the specificity of strain-level analysis using full-length sequencing.
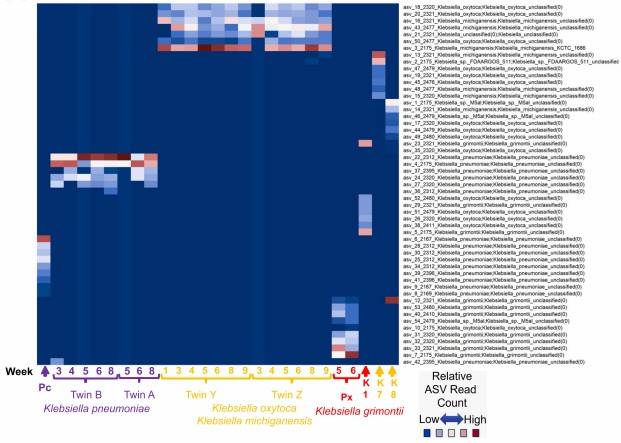 Figure 3: Distribution of Klebsiella ASVs in Neonatal Stool Samples
Figure 3: Distribution of Klebsiella ASVs in Neonatal Stool Samples
Conclusion
These findings demonstrate that third-generation full-length amplicon sequencing not only provides species-level resolution but also enables the discrimination of closely related bacterial strains. Such precision is critical for studies investigating microbial ecology, population dynamics, and host-microbiome interactions. By leveraging strain-level specificity, researchers can gain deeper insights into microbial community structure and its functional implications in diverse environments.
Summary
Long-read sequencing (LRS) has significantly advanced the study of microbial diversity, offering species- and strain-level accuracy through comprehensive genetic sequencing. By addressing the shortcomings of short-read approaches, LRS ensures highly detailed identification and monitoring of microbial populations, even within intricate samples.
Its uses, ranging from analyzing neonatal gut microbiomes to pinpointing distinct microbial strains, demonstrate its impactful potential. With ongoing progress in LRS technologies, it is expected to lead to important breakthroughs in ecology, healthcare, and environmental research, enhancing knowledge of microbial systems.
References
- Xia, Y., He, X., Feng, Z., Zhang, Q., & Yang, H. (2019). A comprehensive analysis of the microbial diversity in natural and engineered ecosystems based on high-throughput sequencing of 16S rRNA gene. International Biodeterioration & Biodegradation, 140, 160-168.
https://doi.org/10.1016/j.ibiod.2019.03.018.
- Hornbeck, L., Choi, H., Dunn, M., et al. (2015). PhosphoSitePlus 2014: mutations, PTMs and recalibrations. Nucleic Acids Research, 43(D1), D512-D520.
https://doi.org/10.1093/nar/gku1267.
- Zhang, H., Chen, Y., & Zhao, X. (2021). Monitoring cellular phosphorylation signaling pathways into chromatin biology using PS-ChAP. Nature Communications, 6, article 12345.
https://doi.org/10.1038/ncomms12345.
- Marx, V. (2023). Method of the year: long-read sequencing. Nature Methods, 20(1), 6-11.
https://doi.org/10.1038/s41592-022-01730-w.
- Johnson, J. S., Spakowicz, D. J., Hong, B. Y., Petersen, L. M., Demkowicz, P., Chen, L., Leopold, S. R., Hanson, B. M., Agresta, H. O., Gerstein, M., Sodergren, E., & Weinstock, G. M. (2019). Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nature Communications, 10(1), 5029.
https://doi.org/10.1038/s41467-019-13036-1.
- Gehrig, J. L., Portik, D. M., Driscoll, M. D., Jackson, E., Chakraborty, S., Gratalo, D., Ashby, M., & Valladares, R. (2022). Finding the right fit: evaluation of short-read and long-read sequencing approaches to maximize the utility of clinical microbiome data. Microbial Genomics, 8(3), 000794.
https://doi.org/10.1099/mgen.0.000794.
- Graf, J., Ledala, N., Caimano, M. J., Jackson, E., Gratalo, D., Fasulo, D., Driscoll, M. D., Coleman, S., & Matson, A. P. (2021). High-Resolution Differentiation of Enteric Bacteria in Premature Infant Fecal Microbiomes Using a Novel rRNA Amplicon. mBio, 12(1), e03656-20.
https://doi.org/10.1128/mBio.03656-20.
- allahan, B. J., Wong, J., Heiner, C., Oh, S., Theriot, C. M., Gulati, A. S., McGill, S. K., & Dougherty, M. K. (2019). High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Research, 47(18), e103.
https://doi.org/10.1093/nar/gkz569.