Overview of EM-seq: A New Detection Approach for DNA Methylation
DNA methylation, a form of chemical modification of DNA, can effect the genetic expression without changing the DNA sequence. DNA methylation plays an important role in the maintenance of the function of normal cells, inactivation of X chromosome in female, stabilization of genome structure, embryonic development, development and progression of certain diseases such as tumer. EM-seq (Enzymatic Methyl-sequencing) is a powerful technique that has been developed to study DNA methylation patterns with high precision and sensitivity.
What is EM-seq
- EM-sep, based on enzymatic treatment of DNA, utilizes specific enzymes that can selectively modify or protect methylated cytosines. The process usually involves in the isolation of genomic DNA from a sample, which can be a cell, tissue, or even body fluids. The isolated DNA is then subjected to enzymatic reactions. One of the key enzymes is methyltransferase, which can add a methyl group to unmethylated cytosines. Then there follows by bisulfite conversion, a process that converts unmethylated cytosines to uracils while the methylated cytosines remain unchanged.
- The advantage of EM-seq compared with traditional bisulfite sequencing (BS-seq) is the improved efficiency and reduced error. In traditional BS-seq, the bisulfite treatment can cause DNA damage, resulting in a lower sequencing quality and accuracy. EM-seq, on the other hand, involves a milder enzymatic method that reduces DNA degradation, therefore induces more accurate detection of methylation sites.
- In addition to the applications in disease research, EM-seq also plays an important role in biological science. It can be used to study the changes in DNA methylation during embryogenesis and cell differentiation. By comparing methylation patterns at different developmental stages, researchers can gain a better understanding of the regulatory networks that control cell differentiation and tissue formation.
Service you may intersted in
For more details, please refer to the following articles:
How EM-Seq Works: A Multi-Step Process
- The experimental method of EM-seq is a multi-step and high-precise process. The first step is the extraction and purification of high-quality genomic DNA from the biological sample. The integrity of the DNA molecule was must be ensured during the extraction process. Once the DNA is obtained, it is subjected to the enzymatic treatment. Here, two main enzymes are used for the protection and cleavage reactions, as described below.
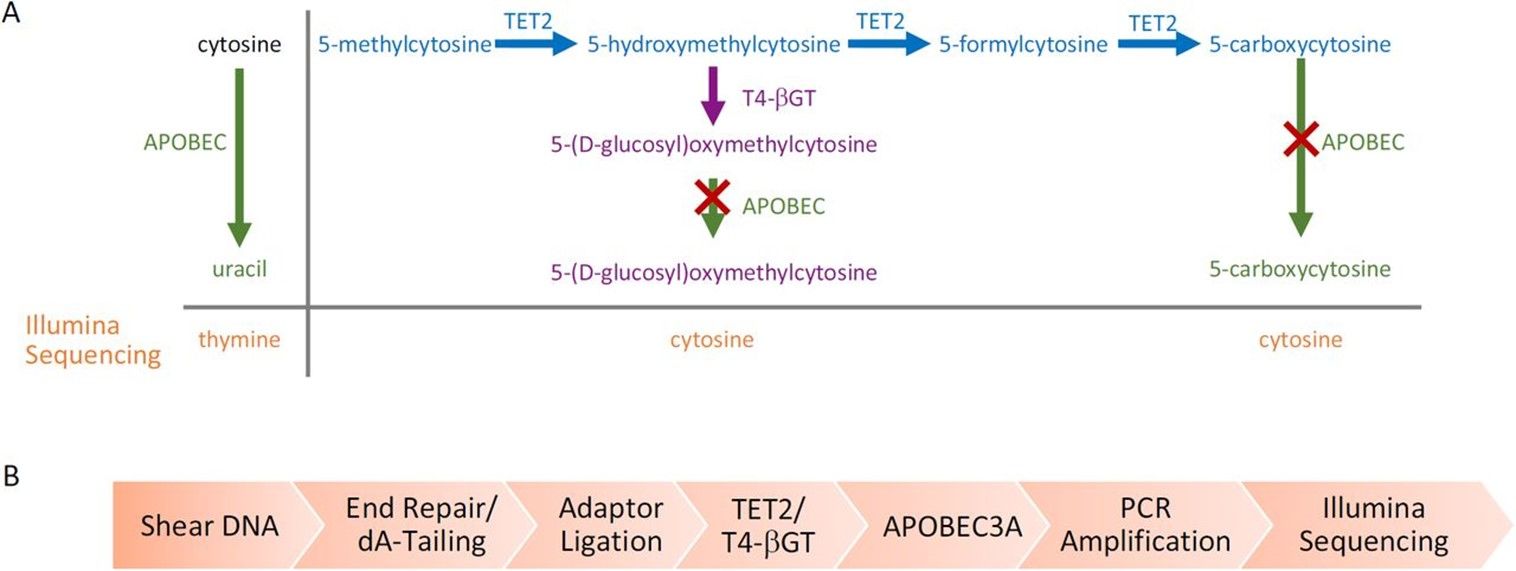 Enzymatic Methyl-seq mechanism of action and workflow. (Romualdas et al., 2021)
Enzymatic Methyl-seq mechanism of action and workflow. (Romualdas et al., 2021)
- Secondly, there are two main types of enzymes are involved in the EM-seq analysis. The first enzyme reaction, which catalyzed by ten-eleven translocation 2 (TET2), could lead to the oxidation of 5-methylcytosine (5-mC) and 5-hydroxymethylated (5-hmC) to 5-carboxylcytosine (5-caC). While the second enzyme reaction, which catalyzed by apolioprotein B mRNA-editing enzyme catalytic polypeptide (APOBEC), could lead to cytosine deamination. However, APOBEC does not affect 5-caC, so 5-mC and 5-hmC could be detected. This step attaches specific DNA sequences to the ends of the fragmented DNA, which is essential for subsequent amplification and sequencing. The amplification step is carried out under controlled conditions to ensure that the desired fragments are amplified successfully.
Enzymes involved in the EM-seq to detect 5-mC and 5-hmC (Romualdas et al., 2021)
- Finally, the amplified library is sequenced using advanced high-throughput sequencing platforms, generating vast amounts of sequence data that can be analyzed to determine the methylation patterns.
How to Analyze EM-seq Data
Quality control: The data generated by EM-seq is both large and complex, and therefore requires a sophisticated data analysis method. Quality control is the first and most important step in the pipeline. The process involves assessing the quality of DNA methylation, checking for any existing contaminants, and ensuring that the data is suitable for further analysis. FastQC can be used to check for length distribution, GC content, and the presence of suitable sequences. Low-quality reads and those with contamination are removed using specific programs such as Trimmomatic.
Methylation Calling: After quality control, the information is recorded onto a reference genome. This recording process requires powerful bioinformatics algorithms and significant computational resources. Once the information recording is complete, methylation response could be made. Methylation response means to distinguish the methylated and unmethylated cytosines based on specific patterns and signals in the sequencing data. Software such as Bismark could be used to identify methylated cytosine sites. The level of methylation at each site can be quantified and is calculated as the ratio of methylation at a particular cytosine site to the total number of methylations at that site.
Differential methylation analysis: Differential methylation analysis is then performed to compare methylation patterns under experimental conditions. This can help in identifying regions of the genome where methylation changes are associated with specific biological processes or diseases. Plenty of tools could be used to analyse methylation, which detects the methylation levels to identify regions that are differentially methylated.
Visualization: Tools like Integrative Genomics Viewer (IGV) could be used to analyse the methylation data of the genome. This allows researchers to analyse the methylation patterns based on genomic structure, including genes, exons, introns, and other genomic features. Visualization of differential methylation levels at specific site can provide deeper insights into the data, and therefore produce insightful interpretation.
Advantages of EM-seq
High resolution: EM-seq can provide single-base resolution of DNA methylation, which allows for a more precise identification of methylation sites on the DNA. For example, EM-seq could be used to precisely detect methylated cytosines when studying gene-regulatory substance, and to gain insights into how methylation affect gene expression even at low DNA concentration.
Reduced DNA damage: Compared to traditional BS-seq, EM-seq employs a milder enzymatic digestion method. While bisulfite treatment in BS-seq could cause DNA damage, the enzymatic digestion process in EM-seq reduce this damage, resulting in DNA with a higher quality for sequencing. EM-seq better protects the integrity of the DNA, resulting in higher accuracy and more reliable detection of methylation sites.
Compatibility with high-throughput sequencing: EM-seq enables the analysis of a large number of samples within a relatively short time. EM-seq, with the characteristic of high-throughput, is crucial for genome-wide methylation studies, where thousands or even millions of methylation sites need to be analyzed of massive samples. For instance, in large-scale cancer epigenetics studies, researchers can quickly analyse a cohort of tumor and normal tissue samples to identify methylation differences associated with the disease by introducing EM-seq during the detection process.
Improved sensitivity and specificity: The enzymatic reactions in EM-seq can offer more accurate identification of methylated and unmethylated cytosines. The specificity of the enzymatic steps and the subsequent analysis can lead to a reduction in false-positive and false-negative results. EM-seq with improved sensitivity and specificity is beneficial for studies that require a high degree of precision, such as those aiming to identify rare methylation events or small methylation changes that could have significant biological implications.
Limitation of EM-seq
Complex enzymatic reactions: The dependence of enzymatic processes in EM-seq means that the technique is more sensitive to enzyme performance and reaction conditions. The enzymes used in the process need to be carefully optimized in terms of temperature, pH, and reaction times. Any change of the conditions, no matter how small it is, could have a great influence of the accuracy and reproducibility of the results. For example, if the temperature is not properly controlled during the methylation-protection step, it could lead to incomplete protection of methylated cytosines and incorrect methylation detection.
Higher cost: The use of specific enzymes and the need for high-quality reagents for EM-seq could make it a relatively expensive technique. Additionally, the high-throughput sequencing also increases the cost. The expense may limit the usage of EM-seq in some laboratories with budget constraints, especially when large-scale studies involving numerous samples are planned.
Bioinformatics challenges: Analyzing EM-seq data requires specialized bioinformatics tools and professional knowledge. The data analysis process is more complex compared to some other sequencing methods due to the specificity of the enzymatic modifications and the need to accurately detect methylation sites. Interpreting the results also requires a good understanding of biological knowledge, any error in the bioinformatics analysis could lead to wrong identification of methylation patterns and therefore draw an incorrect conclusion.
Research Advances in EM-seq
- A recent study published in Genome Research in 2021 shows that EM-seq has higher capabilities of even with very low DNA concentration. The researchers show that EM-seq could accurately detect DNA methylation at single-base resolution with as little as 100 pg of DNA. When compared to BS-seq, EM-seq show more GC distribution, improved correlation between different samples, and the number of CpGs is also increased. These characteristics make EM-seq highly suitable for analyzing samples with limited DNA concentration, such as cell-free DNA (cfDNA) in liquid biopsies or single cells, providing new insights for non-invasive cancer diagnostics and single-cell epigenomics studies.
- In addition, EM-seq palys a more significant role than BS-seq in a variety of specific measurements, including biological analysis. EM-seq can detect approximately 15% more methylation sites than bisulfite methods, providing a more comprehensive view of the methylome. The enzymatic conversion in EM-seq also allows for better DNA integrity, which is important for analysis and obtaining reliable results, especially when working with precious samples.
- Another important development is the combination of EM-seq with other omics data, such as RNA-seq, ATAC-seq, and histone modification data. This approach provides a more comprehensive understanding of the regulatory networks concerning gene expression and cellular function. By integrating these different information, researchers can gain deeper insights into how methylation modifications interact with other molecular to regulate biological processes and how these interactions are disrupted in diseases.
- Despite the many advantages of EM-seq, there is no doubt that it still presents challenges. One of the main challenges is to ensure the enzymatic reactions is complete and accurate. Any mistakes in this process could lead to incomplete modification, which could affect the accuracy of the methylation detection. External contamination, such as residual enzymes or other DNA molecules, could also introduce errors into the analysis. In addition, the interpretation of EM-seq data requires advanced bioinformatics skills and a deep understanding of biological activities. The complexity of the data and the need to consider multiple variables involved make the analysis a complex and time-consuming task.
In conclusion, EM-seq represents a significant milestone in the field of the detection of DNA methylation. Its enzymatic-based approach offers a more accurate, less damaging, and potentially more cost-effective alternative to traditional BS-seq for analyzing DNA methylation patterns. With wide applications in developmental biology, EM-seq is considered to continue making more contributions to our understanding of DNA methylation. However, continuous efforts are also needed to address the technical challenges and improve the data analysis methods to fully realize the potential of this powerful technique.
Reference
- Romualdas V, Zhiyi S, Bradley WL, et al. Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Research, 11.23, (2021): 1280-1289. https://www.genome.org/cgi/doi/10.1101/gr.266551.120.
! For research purposes only, not intended for clinical
diagnosis, treatment, or individual health assessments.