





We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
EM-seq (Enzymatic Methyl-sequencing), a cutting-edge technique for identifying DNA methylation profiles, is showing great potential in various fields, including epigenetic research, disease diagnosis, and the early detection of cancer. Compared to the traditional whole-genome bisulfite sequencing (WGBS), EM-seq offers several advantages. It allows for the creation of DNA sequencing libraries with superior quality, requires less DNA to start with, and results in longer, more intact DNA fragments within the libraries. As technology advances and research deepens, EM-seq is poised to play a more pivotal role in the realms of precision medicine and tailored therapeutics. By providing more accurate and reliable methylation information, it stands to enhance disease prevention and treatment strategies.
Service you may intersted in
For more information, please refer to the following articles:
Researchers, in a study published in the "Clinical Epigenetics" journal, employed EM-seq technology to scrutinize 241 hepatocellular carcinoma (HCC) samples, 76 liver disease samples, and 279 normal plasma samples, thereby evaluating the methylation levels at 1,595 CpG sites. The study pinpointed 283 CpG sites exhibiting significant differences in methylation levels between HCC and non-HCC samples. Based on these markers, a screening model for HCC was constructed, which could proficiently discriminate HCC samples from non-HCC samples, attaining an area under the curve of 0.957 in the test set (with a sensitivity of 90% and a specificity of 97%).
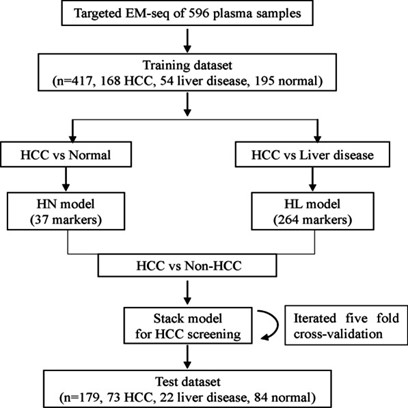 Workflow chart of building a stack HCC screening model. (Guo, Ping et al,2023)
Workflow chart of building a stack HCC screening model. (Guo, Ping et al,2023)
In a recent study, researchers collected plasma samples from 143 individuals with chronic total coronary occlusion (CTO) and conducted EM-seq experiments to analyze DNA methylation patterns. Utilizing the Rendrop CCC grading criteria, patients were stratified into two groups: those with poor collateral circulation (CCC), classified as grades 0 and 1 (34 patients), and those with good CCC, classified as grades 2 and 3 (109 patients). An unsupervised principal component analysis (PCA) of the EM-seq data identified 15 principal components (PCs) that exceeded the noise threshold, with PCs PC1, PC3, and PC8 showing significant correlations with CCC status (p < 1E-4). Notably, PC1 demonstrated a distinct clustering pattern, with samples from patients with good CCC being more dispersed compared to those with poor CCC. In contrast, PC3 revealed minimal overall differences, though a few outliers were observed among the poor CCC group. PC8 indicated a slight trend where good CCC samples were modestly lower than poor CCC samples, offering a degree of discrimination, albeit less pronounced than PC1.
The unsupervised analysis suggested a correlation between methylation patterns in cell-free DNA (cfDNA) from CTO patients and their CCC status, with differential methylation being detected. The researchers then identified differentially methylated regions (DMRs) with q values less than 0.05 and |z| greater than 2 as potential predictors of good CCC. A higher number of downregulated methylation sites were observed compared to upregulated sites. To mitigate the risk of overfitting, DMR analyses were conducted on three resampled subsets of CCC-related DMRs, consistently showing a predominance of downregulated sites. The most consistently reproducible DMRs across these subsets were selected, resulting in 1438 intersecting DMRs. Focusing on the top 500 DMRs from each subset led to the identification of 256 intersecting DMRs. Pathway analysis of these 256 DMRs revealed significant enrichment in pathways associated with TGF-beta, G-protein, and eosinophil biology. PCA using these DMRs effectively discriminated between good and poor CCC in both the training (poor CCC: n = 29; good CCC: n = 93) and test sets (poor CCC: n = 5; good CCC: n = 16), indicating a predictive capability for poor CCC status.
Employing a machine learning approach with the random forest algorithm, the researchers trained a classification model using the 256 DMRs as input features. The model demonstrated high efficacy, with an area under the receiver operating characteristic (ROC) curve of 0.962 in the training set and 0.950 in the test set. The top 20 DMRs, selected based on their "Importance" score, were proposed as CCC markers. Methylation proportion box plots for these markers showed lower levels in good CCC compared to poor CCC, with good CCC exhibiting a broader range and poor CCC being more concentrated near 1. These 20 CCC marker DMRs included 5 exonic, 8 intronic, and 7 intergenic regions. Pathway analysis of these DMRs also indicated significant enrichment in TGF-beta-related pathways, further highlighting the potential role of these regions in CCC biology.
The critical role of early detection in breast cancer screening is paramount for improving patient outcomes and survival rates. While conventional imaging techniques such as mammography and ultrasound are widely used for the early identification of breast cancer, they are not without their limitations, particularly in terms of a high false-positive rate. This can lead to a significant number of unnecessary biopsies, especially among patients classified as BI-RADS 4. Recently, ctDNA, which contains DNA methylation information, has emerged as a promising non-invasive approach for cancer detection, offering new possibilities for the early screening of breast cancer.
In a collaborative research initiative, a team of investigators developed a non-invasive early screening method for breast cancer by conducting epigenetic analyses on ctDNA extracted from liquid biopsy plasma samples. This prospective, multicenter study utilized whole-genome bisulfite sequencing data to assess the clinical utility of cfDNA methylation markers in 203 women presenting with suspected malignant breast lesions. Remarkably, this approach significantly improved the early diagnosis of BI-RADS 4 patients, as indicated by an increase in the area under the curve (AUC) from 0.78 to 0.79 to a more robust 0.93 to 0.94. This study marks the first blood-based whole-genome DNA methylation investigation capable of detecting early breast cancer with single-base resolution, distinguishing it from benign tumors. It thus highlights the potential of integrating liquid biopsies with traditional diagnostic imaging to reduce false-positive rates and prevent unnecessary medical interventions.
This research underscores the importance of innovative approaches in breast cancer screening, demonstrating how the integration of advanced molecular techniques with established imaging modalities can enhance diagnostic accuracy. The findings suggest that the incorporation of ctDNA methylation analysis into clinical practice could lead to more precise patient stratification and personalized treatment strategies, ultimately contributing to better patient management and improved survival rates in breast cancer.
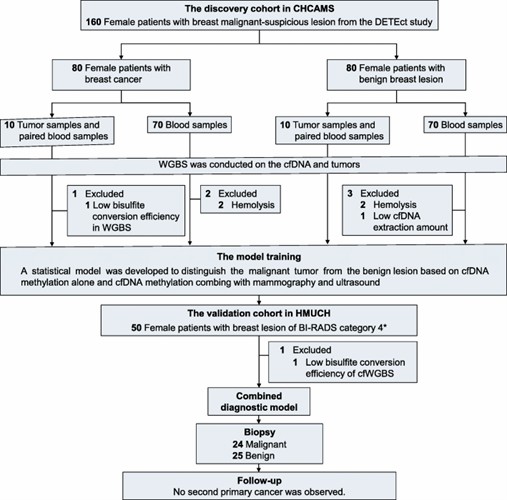 Experimental Approach. (Liu, J., Zhao, H., Huang, Y.et al.2021)
Experimental Approach. (Liu, J., Zhao, H., Huang, Y.et al.2021)
MESA technology is an innovative approach that integrates various epigenetic features of ctDNA, including methylation patterns, nucleosome occupancy, and nucleosome phasing. This comprehensive analysis significantly enhances the accuracy and depth of cancer detection. Unlike traditional methods that rely on bisulfite treatment, MESA employs techniques such as Enrichment of Methylation through EM-seq or Targeted Amplicon Pyrosequencing (TAPS) to perform whole-genome methylation profiling, which allows for the early detection and classification of tumors.
In two independent colorectal cancer clinical cohorts, MESA demonstrated high concordance or superiority over conventional methods by conducting targeted EM-seq sequencing on 462 ctDNA samples. The multimodal strategy of MESA has shown increased accuracy in detecting colorectal, liver, and pancreatic cancers compared to single-modal models, highlighting the importance of leveraging multimodal epigenetic signatures for non-invasive cancer detection.
The advancement of MESA represents a significant milestone in the field of non-invasive cancer detection, with the potential to refine clinical protocols by improving diagnostic precision and reducing unnecessary medical interventions. Future studies are necessary to validate MESA's findings in larger sample sizes and across multiple centers, as well as to assess its clinical applicability and stability in identifying subtype-specific methylation biomarkers. This research direction is poised to contribute to the evolution of personalized medicine and enhance our ability to detect and manage cancer more effectively.
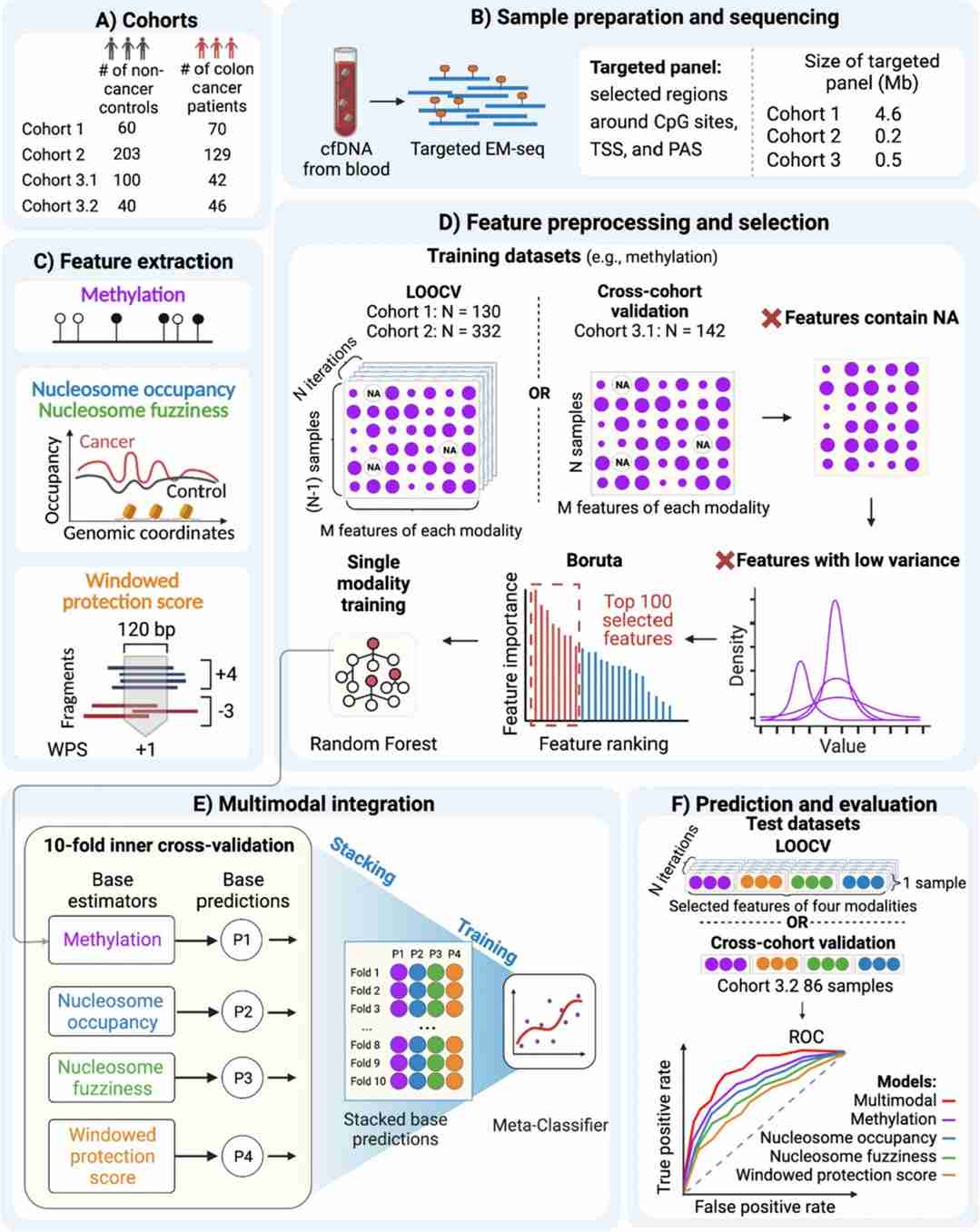 Schematic diagram displaying the design of MESA. (Li, Y., Xu, J., Chen, C.et al.2024)
Schematic diagram displaying the design of MESA. (Li, Y., Xu, J., Chen, C.et al.2024)
The future application potential of EM-seq technology is primarily reflected in the following aspects:
Elevated Precision and Productivity in DNA Methylation Detection: EM-seq is capable of discerning methylation at the single-base resolution from minute amounts of DNA, even in picogram quantities. This proficiency is essential for scenarios demanding low-input DNA, like low-abundance cfDNA or deteriorated formalin-fixed paraffin-embedded (FFPE) specimens.
Minimization of DNA Impairment and Distortion: In contrast to conventional bisulfite sequencing, EM-seq utilizes enzymatic reactions rather than chemical processes, which curtails DNA damage and fragmentation. Consequently, it lessens bias and augments data fidelity.
Enhancement of GC Content Bias: The EM-seq technology affords more consistent coverage across regions with diverse GC content. This is especially vital for investigating methylation patterns in GC-rich regions, which are recognized to have a pivotal role in cancer progression and monogenic disorders.
Expansion of Application Domains: EM-seq is not only suitable for library construction on Illumina platforms but also for target enrichment, reduced representation libraries, or amplicon detection, among other uses. Such flexibility enlarges the scope of EM-seq in a broader spectrum of research and clinical applications.
Promoting Precision Medicine and Personalized Treatment: By furnishing more accurate and comprehensive methylation data, EM-seq is highly beneficial for the progress of precision medicine and personalized therapy. It facilitates early disease diagnosis and treatment response monitoring by more precisely detecting disease-related methylation alterations.
Diminution of Sequencing Expenses: EM-seq yields more utilizable sequencing data from an equivalent number of reads, which can ultimately reduce sequencing costs and render a wider variety of research and clinical applications practicable.
In conclusion, EM-seq technology, with its distinctive merits in DNA methylation detection, exhibits colossal potential in precision medicine, personalized therapy, and numerous research applications. As the technology persists in advancing and being implemented, EM-seq is anticipated to assume an increasingly prominent position in forthcoming epigenetic research and clinical practice.
References
Terms & Conditions Privacy Policy Copyright © CD Genomics. All rights reserved.