






We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
DNA methylation is a form of DNA chemical modification that can regulate genomic function without altering the primary structure of DNA, causing changes in chromatin structure, DNA stability, and DNA protein interaction patterns, thereby affecting gene expression.
The so-called DNA methylation refers to the covalent bonding of the cytosine 5 'carbon position of CpG dinucleotide in the genome to a methyl group under the action of DNA methyltransferase. Numerous studies have shown that DNA methylation is closely related to physiological and pathological processes such as embryonic development [1], genomic structural stability [2], genetic imprinting [3], aging [3], and the occurrence and development of tumors and diseases [4], playing important biological functions in animal and plant life activities. DNA methylation plays a decisive role in the maintenance, self-renewal and differentiation of stem cells, aging and developmental abnormalities of individuals, and the occurrence and development of diseases (such as tumors, diabetes, psychosis, nervous system diseases and other complex diseases). With the continuous innovation of global DNA methylation analysis technology, our understanding of genomic DNA methylation in various aspects such as physiology and pathology has been expanded.
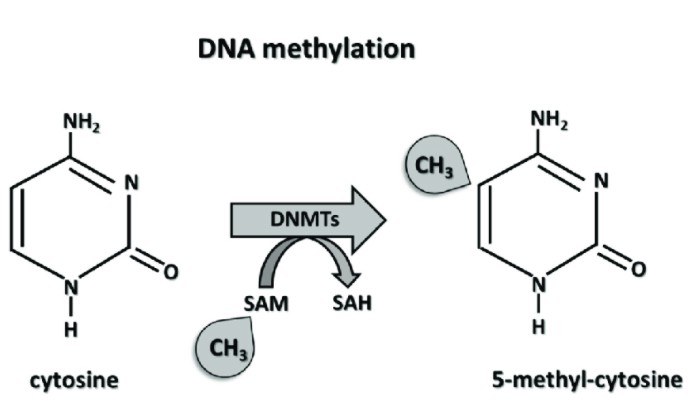 The mechanism of DNA methylation.Marzena (Ciechomska et al,. 2019)
The mechanism of DNA methylation.Marzena (Ciechomska et al,. 2019)
The most extensively studied form of DNA methylation modification currently is 5-mC, which is widely present in the genomes of eukaryotes such as plants and animals, and is known as the "fifth base" of DNA. 5-hmC, on the other hand, is known as the "sixth base" of the mammalian genome and plays an important role in the occurrence and development of diseases such as the nervous system and cancer [6,7]. To learn more about DNA methylation, refer to "What is DNA Methylation"
In the past decade, DNA methylation has always been a key direction of funding from the fund. Searching with the keyword "DNA methylation", it was found that the number of articles related to DNA methylation is still considerable; Using "DNA methylation seq" as the keyword search, it was found that the number of articles related to DNA methylation sequencing is increasing year by year, and research hotspots mainly focus on animal and plant, microbiology, diseases, and cancer.
With the development of high-throughput sequencing technology, we are able to analyze 5 '- methylcytosine at the whole genome level, which is called "DNA methylation sequencing". In recent years, with the continuous decrease in sequencing costs and the iterative updates of sequencing technology, DNA methylation sequencing methods have become more selective.
In recent years, advancements in sequencing technologies have revolutionized our ability to study DNA methylation at a genome-wide scale. This article provides a comprehensive overview of various DNA methylation sequencing methods, their principles, technical features, and applications.
At present, there are five common sequencing methods for epigenetic DNA methylation research, including whole genome bisulfite methylation sequencing (WGBS), Reduced Representation Bisulfite Sequencing (RRBS), methylated DNA immunoprecipitation sequencing (MeDIP seq), methylation capture sequencing/Targeted Methylation Sequencing (Methyl-Seq), and methylation chip detection.
Service you may intersted in
It can accurately detect Gthe methylation levels of all individual cytosine bases (C bases) across the entire genome. Can detect the methylation levels of all loci in the entire genome; It is a relatively early and universal detection technique.
Conventional whole genome methylation sequencing technology uses T4-DNA ligase to break the connector sequence at both ends of genomic DNA fragments by ultrasound. The connector product is transformed from unmethylated cytosine C to uracil U through bisulfite treatment, and then transformed from uracil U to thymine T through connector sequence mediated PCR technology to distinguish whether the cytosine C base has undergone methylation.
Technical advantages:
Whole genome coverage: Maximizing the acquisition of complete genome wide methylation information;
Single base resolution: can accurately analyze the methylation status of each C base.
Technical defects:
The price is expensive, the cost-effectiveness is not high, and it is not suitable for making a large number of samples;
he sequencing depth is low, and the average depth of 90G data for human samples is 12-15X.
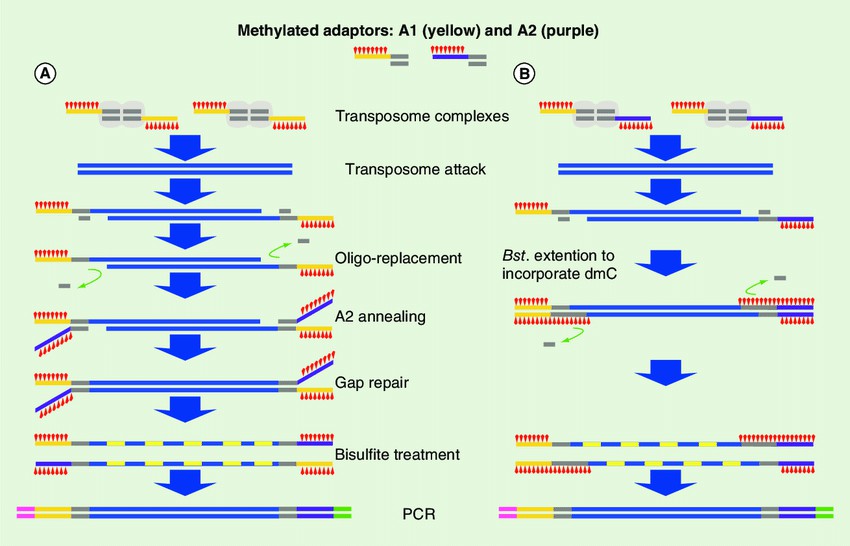 The modified tagementation-based whole genome bisulfite sequencing method. (Hanlin Lu et al,.2015)
The modified tagementation-based whole genome bisulfite sequencing method. (Hanlin Lu et al,.2015)
RRBS is the use of restriction endonucleases to cleave the genome, enrich regions with relatively concentrated CpG sites, and perform heavy sulfite sequencing. This technology improves the sequencing depth of high CpG regions, mainly achieving high-precision resolution in CpG islands and regions.
Technical advantages:
1.Single base resolution: Within its coverage range, it can achieve single base resolution;
2.High cost-effectiveness: The sequencing area targets high CpG areas, resulting in higher data utilization.
Technical defects:
1.Low repeatability: The repeatability of the technology is only about 85% -95%;
2.Sequencing depth: A data volume of 10g, with an average sequencing depth of 30X. The proportion of sites with sequencing depth greater than 30X is relatively low.
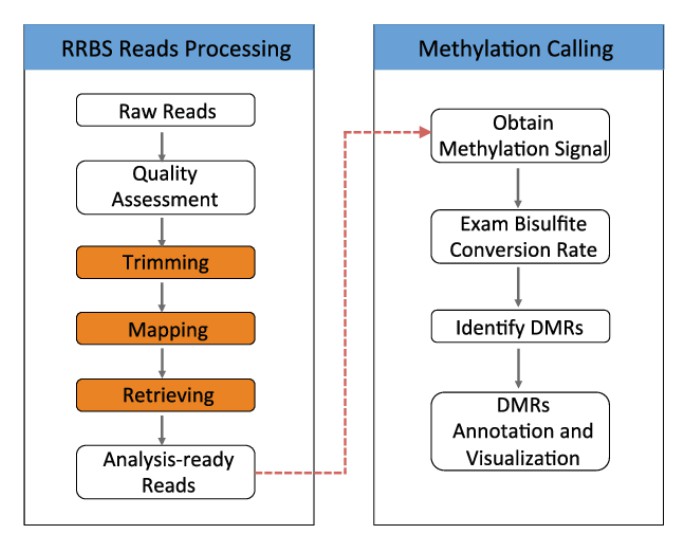 Schematic view of the RRBS sequencing analysis workflow. (Xuefeng Wang et al,.2015)
Schematic view of the RRBS sequencing analysis workflow. (Xuefeng Wang et al,.2015)
MeDIP Seq is a genome-wide methylation detection technology based on the principle of antibody enrichment. It uses methylated DNA immunoprecipitation technology to specifically enrich DNA fragments that undergo methylation on the genome through 5 '- methylcytosine antibodies. Then, high-throughput sequencing can be used to study high-precision CpG dense high methylation regions at the genome-wide level.
Technical advantages:
Low cost: Significantly reduces the amount of sequencing data (only 1-2G per sample), resulting in low sequencing costs;
Technical defects:
Single base resolution: unable to detect methylation values of individual sites, with a resolution of 50-100bp.
Methyl Seq is a targeted methylation detection technique that uses probe capture principles for sequencing. The experimental principle is similar to whole exome sequencing, and high-throughput sequencing can be used to study high-precision CpG dense high methylation regions at the genome-wide level. The designed intervals are at the genome-wide level, including CpG islands, CpG island beaches, CpG island shelves, and unmethylated regions Promoter, tumor and tissue specific methylation region (DMR), enhancer, Ensemble regulatory region, and DNase I high sensitivity site.
Technical advantages:
1.Single base resolution: Within its coverage range, it can achieve single base resolution;
2.Sequencing depth: The sequencing depth can reach 80-100x when measured at 15G, with high cost-effectiveness;
3.High repeatability: Technical repeatability correlation R2>0.97;
4.Can perform methylation haplotype analysis;
5.The methylation status is not affected by SNP sites;
6.Flexible design of different panels according to customer needs.
Technical defects:
At present, product-based targeted methylation sequencing products can only detect humans, rats, and mice.
Illumina methylation chip detection
The Illumina Methylation Chip is a chip designed based on the principle of chip hybridization to capture single base extension. It has been upgraded from the initial 27K, 450K, and 850K to the 935K chip. The chip has also been continuously improved and upgraded to the currently considered optimal version. The methylation chip is also a product that covers the entire genome, covering CpG islands, promoters, coding regions, open chromatin, and enhancers. In addition, it also includes CpG sites outside the CpG island, known DMR sites, deoxyribonuclease hypersensitivity sites, and miRNA promoter regions.
Technical advantages:
1.Single base resolution: Within its coverage range, it can achieve single base resolution;
2.High repeatability: Technical repeatability correlation R2>0.99;
3.There are a large number of public databases available for utilization.
Technical defects:
Not suitable for analysis of methylation segments.
Epigenetic modifications do not require changes in DNA sequence to achieve changes in traits, and these changes are closely related to gene function, cellular status, development, aging, diseases, and more. Among numerous epigenetic modifications, one of the most important and widely studied is DNA methylation, and whole genome methylation sequencing (WGBS seq) is undoubtedly the most effective research method.
Whole genome methylation sequencing utilizes the property of bisulfite to convert unmethylated cytosine (C) into thymine (T). After treating the genome with bisulfite, sequencing can calculate the methylation rate based on the ratio of the number of reads on a single C site that were not converted to C but not to T to the number of reads covered. This technology has important application value for comprehensive research on embryonic development, aging mechanisms, epigenetic mechanisms of disease occurrence and development, as well as screening for disease related epigenetic marker sites.
The overall process of whole genome methylation sequencing technology can be divided into five parts, namely sample preparation, genomic DNA extraction, library construction, bisulfite treatment, machine sequencing, and data analysis
DNA methylation exerts a significant impact on embryonic development by determining cell fate, facilitating tissue differentiation, and supporting the formation of organs—organogenesis. Throughout embryogenesis, genes' expression is precisely regulated by fluctuations in DNA methylation patterns, thereby fine-tuning the multitude of developmental processes. Technological advancements such as whole-genome bisulfite sequencing (WGBS) provide unprecedented insights into the fluid epigenetic landscape that defines embryonic development. These technologies aid in identifying crucial regulatory zones and the biological pathways that guide cellular lineage selection and morphogenesis.
DNA methylation, a pivotal component of epigenetic regulation, undergoes extensive alterations as an organism progresses through its lifespan. These modifications exert a substantial influence on aging processes, playing a significant role in the onset and progression of diseases associated with advancing age. Epigenetic clocks based on DNA methylation, in providing accurate estimations of chronological age, illuminate the expansive biological intricacies inherent to the process of aging. Research endeavors, utilizing methylation sequencing methodologies, have yielded fascinating insights, exposing age-related alterations across genomic landscapes, inclusive of promoters, enhancers, and repetitive elements. The amassed evidence implicates, with notable weight, the role of epigenetic dysregulation in the pathogenesis of a spectrum of age-related conditions, spanning from neurodegenerative disorders and heart-related afflictions to various malignancies.
DNA methylation disturbances play a significant role in cancers, contributing largely to the initiation, progression, and metastasis of tumors. Unique tumor-specific DNA methylation patterns serve as powerful diagnostic and prognicative tools that guide detailed patient categorization and evidence-based therapeutic decisions in oncology. A spectrum of methylation sequencing techniques, exemplified by targeted methods such as Methyl-Seq and comprehensive genome-wide strategies like WGBS, enables a comprehensive exposition of the cancer methylome panorama. Consequently, these approaches facilitate the detection of novel epigenetic aberrations and potential therapeutically targetable loci. Furthermore, the emergence of epigenetic therapies that target DNA methylation modifiers, encompassing DNA methyltransferase inhibitors and histone deacetylase inhibitors, represents a promising frontier in precision oncology and customized cancer treatment paradigms.
Multiple environmental determinants, including various pollutants, dietary habits, and lifestyle choices, hold potential for altering DNA methylation profiles. These alterations can potentially impact predispositions towards diseases, thereby affecting overall health trajectories. The intricate field of environmental epigenetics seeks to unravel the complex relationship between environmental triggers and epigenetic modifications, thereby offering a molecular perspective on the dynamic interaction between genes and the environment. Empirical investigations employing methylation sequencing approaches have unveiled specific CpG sites that respond to environmental cues, notably those related to the exposure to toxins, pollutants, and diverse stress factors. Insights garnered from these investigative revelations enhance our understanding of the etiological mechanisms underlying environmentally-driven diseases such as asthma, diabetes, and cardiovascular disorders.
Within the rapidly evolving field of DNA methylation sequencing, a myriad of technological and systematic advances continuously drive substantial transformations in established principles and techniques. Emerging methodologies encompassing single-molecule sequencing, nanopore sequencing, and spatial profiling at a highly resolved scale hold promise in overcoming previous limitations and fostering novel breakthroughs in methylation sequencing. These innovative strategies, characterized by enhanced resolution, sensitivity, and scale of operation, provide an advantageous platform for conducting comprehensive examinations of DNA methylation dynamics traversed by an extensive spectrum of biological contexts.
Assimilation of DNA methylation information with diverse epigenomic, transcriptomic, and proteomic datasets underpins comprehensive multi-omics profiling, thereby offering refined understanding of gene regulation and cellular functions. The integration of such multi-omics methodologies aids in deciphering intricate regulatory networks, uncovering mechanisms driving diseases, and identifying potential therapeutic targets. This amalgamation of approaches presents new trajectories for precision medicine and personalized healthcare. Thus, by coupling methylation sequencing data with allied omics datasets, researchers have the opportunity to unravel the minute molecular details at play in health and disease, offering unprecedented insights.
DNA methylation, as an important epigenetic regulatory mechanism, plays a crucial role in many key biological processes such as development and disease progression. In recent years, the introduction of methylation analysis into early screening of tumor liquid biopsy based on free nucleic acid has pushed the application of DNA methylation to a more hot wave. Compared to traditional chip based detection methods, Whole Genome Bisulfite Sequencing (WGBS) technology based on high-throughput sequencing combines efficient bisulfite conversion with high-throughput sequencing library construction method (Post-BS WGBS), which can accurately analyze methylation sites from a wider range of sample genomes at single base resolution. It can cover all methylation sites and also cooperate with targeted technology to detect low-frequency methylation signals. It has become a mainstream method that has received widespread attention in the industry.
However, as a challenging sequencing method in practical operation, the traditional WGBS library construction method faces many challenges, such as high requirements for initial DNA quantity, cumbersome operation process, long library construction time, and high errors and bias rates introduced by library amplification. With the continuous deepening of its application in the field of cancer, for common samples such as FFPE and ccfDNA with low initial quantity and poor fragment integrity, it will face serious efficiency problems from the early stage of bisulfite transformation, which will seriously affect the quality of subsequent library construction and sequencing.
References
Terms & Conditions Privacy Policy Copyright © CD Genomics. All rights reserved.