Exploring cfDNA Methylation Fundamentals and Its Clinical Relevance in Cancer
- cell-free DNA (cfDNA) methylation serves as a critical biomarker in liquid biopsy research, offering valuable insights into the genetic and epigenetic alterations that characterize tumors. The identification of tumor-specific cfDNA through methylation pattern analysis is a significant tool for early cancer detection, monitoring disease progression, assessing treatment efficacy, and evaluating organ viability in transplantation medicine. Notably, researchers at Tsinghua University have developed a method utilizing cfDNA methylation for early cancer diagnosis, enhancing the detection of tumor-derived cfDNA in patients through the analysis of fragments across diverse datasets.
- Furthermore, cfDNA methylation is not limited to early cancer diagnosis; it also extends its utility to the surveillance of disease progression and the evaluation of treatment efficacy. By analyzing alterations in cfDNA methylation patterns before and after treatment, clinicians can assess treatment success, identify the origins of primary tumors, and predict disease risks. Additionally, the examination of cfDNA methylation profiles reveals the heterogeneity within tumors, encompassing both genomic and epigenetic diversity, which is crucial for understanding the development and invasive behavior of cancers. These capabilities underscore the growing importance of cfDNA methylation in personalized medicine and precision oncology, highlighting its role in tailoring therapeutic strategies to individual patient needs.
What Is cfDNA Methylation?
- The term "cfDNA methylation" refers to the epigenetic modification that takes place in cfDNA. These small DNA fragments circulate in the bloodstream, primarily tilizing cfdna methylation for early cancetracking of disease progressioncovalent addition of a methyl group to cytosine residues, particularly in regions rich in CpG dinucleotides, termed CpG islands. This modification can profoundly influence gene expression and plays a critical role in various biological processes, including cellular differentiation, development, and the progression of numerous diseases.
- The methylation status of cfDNA serves as a critical biomarker in liquid biopsies, offering valuable insights into the genetic and epigenetic alterations occurring within tumors. This process involves the transfer of a methyl group from a donor molecule to the cytosine base, which can modify the transcriptional activity of genes. In cancer, abnormal methylation patterns often indicate tumor development; for example, normally unmethylated CpG islands in gene promoter regions may become hypermethylated, leading to gene silencing. Conversely, hypomethylation can occur within gene bodies, potentially enhancing oncogenic processes.
- Investigating cfDNA methylation is particularly advantageous for cancer diagnostics and monitoring, as it enables the non-invasive identification of tumor-derived DNA in the bloodstream. This approach has the potential to revolutionize early cancer detection, facilitate the tracking of disease progression, and evaluate treatment effectiveness without the need for invasive tissue biopsies. As research continues to elucidate the complexities of cfDNA methylation patterns, its role in personalized medicine and precision oncology is expected to expand significantly.
How Is cfDNA Methylation Analysis Done
This approach typically involves the application of bisulfite treatment, which selectively converts unmethylated cytosines to uracil while leaving methylated cytosines unchanged. Following this modification, the DNA is amplified and sequenced. The preservation of cytosine bases in the sequence data allows for the precise identification of methylated cytosines, thereby revealing the methylation status of the DNA.
Pyrosequencing
Bisulfite treatment and pyrosequencing can be used to quantitatively measure DNA methylation at specific CpG sites. It involves sequencing by synthesis and provides real-time analysis of the incorporation of nucleotides, which corresponds to the methylation status.
Methylation-Specific PCR (MSP)
This method employs primers that are designed to be specific to either the methylated or unmethylated forms of DNA sequences. By conducting separate PCR with each set of primers, researchers can ascertain the methylation status of specific gene promoters or genomic regions. This strategy allows for a clear distinction between the methylated and unmethylated DNA, providing insights into the epigenetic regulation of gene expression.
Microarrays and Next-Generation Sequencing (NGS)
Advanced techniques such as microarray analysis and NGS facilitate the examination of DNA methylation across the entire genome. By leveraging these high-throughput platforms, researchers are able to concurrently evaluate methylation patterns at numerous genomic loci. This comprehensive approach provides an in-depth understanding of the methylome, thereby revealing the collective methylation landscape within a given biological context.
CpG Island Analysis
Special attention is often given to CpG islands, which are regions with a high frequency of CpG sites. Methylation in these regions can have a significant impact on gene expression, and specialized assays target these areas for detailed analysis.
cfDNA arises from a variety of biological processes and sources within the human body. Under normal physiological conditions, cfDNA primarily results from the breakdown of genomic DNA in senescent and apoptotic cells. However, in pathological situations such as cancer, trauma, organ transplant rejection, organ failure, and severe infections, necrotic cells can release substantial amounts of DNA into the bloodstream.
Additionally, during pregnancy, cfDNA can originate from the fetus, with fetal DNA entering the maternal circulation. In the context of organ transplantation, cfDNA may also be released into the blood due to cell death linked to immune rejection. Moreover, cfDNA can be actively secreted by certain cells, although the precise mechanisms underlying this process remain to be fully elucidated.
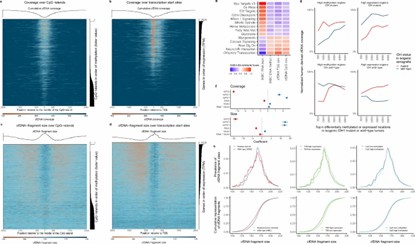 Effect of CpG methylation and gene expression on coverage and size of cfDNA fragments. (Noë, M., Mathios, D., Annapragada, A.V. et al.)
Effect of CpG methylation and gene expression on coverage and size of cfDNA fragments. (Noë, M., Mathios, D., Annapragada, A.V. et al.)
Service you may intersted in
For more information, please refer to the following articles:
cfDNA Methylation Database
- The cfDNA Methylation database is a comprehensive repository that compiles methylation profiles from cfDNA samples of individuals afflicted with diverse cancer types. This collection is amassed through an array of analytical methodologies for methylation, such as bisulfite sequencing, pyrosequencing, MSP, microarray analysis, NGS, and the examination of CpG islands. These sophisticated techniques enable the precise detection and quantification of methylation patterns within specific DNA sequences, thereby affording researchers a more profound insight into the intricacies of tumorigenesis and the dynamics of cancer progression.
- The data contained within the database serve as a valuable resource for scientists aiming to identify and validate methylation markers unique to cancer. These markers hold significant promise for a range of clinical applications, including the early detection of cancer, classification of disease subtypes, assessment of patient prognosis, and monitoring of therapeutic outcomes. For instance, the use of WGBS allows researchers to survey nearly 88% of the human genome with single-base resolution. This approach has elucidated the methylation profiles of cfDNA across various genomic regions and has revealed an inverse relationship between cfDNA quantity in specific regions and their CpG density.
- The cfDNA Methylation Database serves as an indispensable repository for scientists delving into the implications of cfDNA methylation in oncogenesis, as well as for those engaged in the innovation of diagnostic methodologies and therapeutic approaches. Through meticulous examination of the methylation profiles archived within, researchers are equipped to dissect the complexities of tumor heterogeneity and pinpoint potential therapeutic targets for cancer intervention. The establishment and ongoing curation of such databases are pivotal for propelling the frontiers of personalized medicine and the precision oncology movement.
What Are the Common Methods for Detecting cfDNA Methylation
- Detecting DNA methylation, especially within the realms of cancer research and clinical practice, often relies on bisulfite sequencing as a predominant technique. This approach entails the exposure of DNA to sodium bisulfite, which selectively converts unmethylated cytosines into uracil, while cytosines that are methylated remain unaltered. Post-treatment, the DNA undergoes amplification via PCR and subsequent sequencing. The preserved cytosine bases in the sequencing reads indicate the locations of methylated cytosines. Bisulfite sequencing is favored for its capacity to yield intricate methylation patterns with single-base precision across the entire genomic landscape.
- The Illumina Human Methylation BeadChip is a widely recognized microarray platform that facilitates the simultaneous assessment of methylation at numerous CpG sites. This technology is highly valued for its capacity to handle large-scale analyses and for the extensive dataset it can generate. As a result, it has become an essential instrument in the fields of developmental biology and oncology, providing researchers with a robust means to investigate methylation patterns associated with various biological processes and diseases.
- Additionally, NGS platforms are dominant in DNA methylation profiling approaches, offering the advantage of analyzing methylation on a genome-wide scale. These technologies enable the assessment of methylation at numerous loci, providing a comprehensive view of the methylome.
- In conclusion, bisulfite sequencing stands as a foundational technique for identifying DNA methylation, yet the Illumina Human Methylation BeadChip and NGS platforms are equally prevalent. Their high-throughput nature and extensive methylome coverage make them indispensable. These diverse methodologies are vital for deciphering the impact of DNA methylation across a spectrum of biological phenomena and pathologies, with a particular emphasis on oncology.
cfDNA Methylation Quality Control
- cfDNA Methylation Quality Control (CfDNA Methylation QC) is a critical step in ensuring the accuracy and reliability of cfDNA methylation analysis results.
- QC is a critical component in the analysis of cfDNA methylation to safeguard the precision and reliability of the findings. At the outset, rigorous QC protocols must be adhered to throughout the extraction and processing of DNA samples. For example, the integrity of the sequencing outcomes is evaluated by assessing metrics such as the count of uniquely aligned reads, the alignment rate, and the rate of duplicated sequences. Samples are typically disqualified if they contain less than 150 million uniquely aligned reads, exhibit an alignment rate of less than 80%, have a duplication rate exceeding 25%, or if the efficiency of C to T conversion falls below 99%. These criteria are essential to maintain the integrity of the data and ensure that the conclusions drawn from the methylation analysis are scientifically sound.
- In bisulfite sequencing, which is frequently employed for cfDNA methylation analysis, it is crucial to evaluate the efficiency of the bisulfite treatment to confirm that unmethylated cytosines are adequately converted to uracil. Additionally, for microarray platforms like the Illumina Human Methylation BeadChip, quality control encompasses the evaluation of signal intensity, background noise, and the uniformity of hybridization. These assessments are essential to ensure the reliability and accuracy of the methylation data obtained from these techniques.
- In sequencing techniques that utilize affinity enrichment, such as cfMeDIP-seq, quality control involves evaluating both the specificity and efficiency of the immunoprecipitation process. This can be accomplished by incorporating synthetic spike-in DNA controls, which serve as known references for quantitative normalization. These controls help mitigate batch effects and biases associated with the physical characteristics of the DNA fragments, thereby enhancing the reliability of the results.
- In whole-genome methylation sequencing (WGMS), quality control also includes the definition of methylated regions, quantification of methylation levels, and the accuracy of retrieving methylation information from databases such as TCGA. Through these rigorous quality control measures, researchers can ensure the quality and reliability of cfDNA methylation data, thereby providing accurate biomarkers for cancer research and clinical applications.
How Can cfDNA Sequencing Improve Methylation Characterization
High Sensitivity and Specificity
cfDNA methylation sequencing provides a highly sensitive and specific method for identifying cancer-related methylation alterations, making it a highly valuable tool for liquid biopsy applications. Its exceptional sensitivity makes cfDNA sequencing particularly beneficial for the early detection of cancer and the ongoing monitoring of disease progression.
Cost-Effectiveness
The cfMethyl-Seq technology enriches CpG sites, thereby reducing the cost of whole-genome methylation profiling while capturing the majority (>90%) of CpG islands. This approach significantly lowers costs compared to traditional WGBS, while still providing comprehensive methylation data.
Low Input Requirements
Methods such as cfMeDIP-seq and MCTA-seq can analyze methylation from very low DNA input amounts (1-10 ng), which is particularly important for cfDNA, a sample type that is often present in low quantities.
Avoidance of PCR Amplification Bias
The use of nanopore sequencing technology avoids GC bias and DNA damage during PCR amplification, generating up to 200 million reads directly from cfDNA, an order of magnitude improvement over existing nanopore sequencing methods.
Single-Molecule Classifier
The development of a single-molecule classifier to determine whether individual reads originate from tumor or immune cells aids in more precisely characterizing cfDNA methylation features.
The Role of cfDNA Methylation in Cancer Detection and Surveillance
Early Cancer Detection and Diagnosis
Methylation patterns in cfDNA hold significant promise for the early detection and diagnosis of cancer, offering a non-invasive method to assess tumor dynamics and subtypes. Aberrant DNA methylation is among the earliest molecular changes in carcinogenesis and can be detected in various biological samples. By examining these methylation signatures in cfDNA, we can identify biomarkers that reflect the initial molecular alterations in cancer, which is crucial for improving early detection rates. This approach provides a more comprehensive understanding of the cancer's genetic and epigenetic landscape, thereby laying the groundwork for personalized therapeutic strategies and patient management.
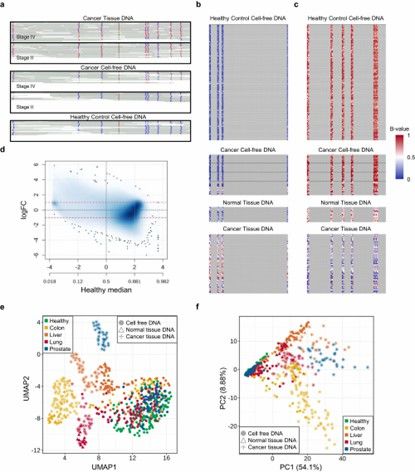 Cancer signature captured in methylation patterns. (Kim, S.Y., Jeong, S., Lee, W.et al.)
Cancer signature captured in methylation patterns. (Kim, S.Y., Jeong, S., Lee, W.et al.)
Monitoring Treatment Response
The examination of methylation patterns in cfDNA emerges as a crucial approach for the continuous monitoring of cancer progression and recurrence. By analyzing changes in cfDNA methylation profiles before and after therapeutic interventions, clinicians can obtain real-time insights into the effectiveness of treatments. This information is invaluable in guiding clinical decisions and adapting treatment strategies to enhance patient outcomes.
In the realm of rectal cancer, research has shown that an increase in LINE-1 methylation levels post-treatment signifies a favorable therapeutic response, whereas modifications in the methylation status of specific genes may indicate recurrence. These observations highlight the potential of cfDNA methylation as a biomarker for assessing treatment efficacy and detecting early signs of cancer relapse, thereby enabling more personalized and timely clinical interventions.
Tumor Heterogeneity and Subtypes
The evaluation of methylation profiles within circulating cfDNA constitutes a pivotal area of investigation in oncology, as it provides a detailed map of tumor heterogeneity and enables the differentiation of distinct tumor subtypes. This analytical approach effectively uncovers the complex genomic and epigenetic variations inherent in neoplasms, offering critical insights into the disease's progression and its potential for metastasis.
In the context of small cell lung cancer (SCLC), the examination of cfDNA methylation patterns has demonstrated significant utility in distinguishing between disease subtypes. This capability is not merely theoretical; it has practical implications, as it supports the development of individualized treatment strategies and facilitates precise patient classification. The ability to accurately identify tumor subtypes is essential for the advancement of personalized medicine, ensuring that therapeutic approaches are both targeted and maximally efficacious.
Continuous Surveillance and Recurrence Forecasting
The analysis of methylation patterns within circulating cfDNA represents a robust approach for monitoring cancer progression and potential recurrence. By systematically tracking cfDNA methylation levels across time, it is feasible to identify early signals of tumor re-emergence, thereby enabling timely therapeutic interventions. This dynamic surveillance strategy not only enhances the sensitivity of early cancer detection but also provides valuable insights into treatment efficacy and the likelihood of disease relapse.
In conclusion, cfDNA methylation has emerged as a pivotal biomarker in cancer diagnostics and longitudinal monitoring. Its application not only augments early-stage detection rates but also supports the evaluation of therapeutic outcomes and the prediction of recurrence, offering a promising avenue for personalized cancer care and precision medicine.
References
- Kim, S.Y., Jeong, S., Lee, W.et al. Cancer signature ensemble integrating cfDNA methylation, copy number, and fragmentation facilitates multi-cancer early detection. Exp Mol Med 55, 2445–2460 (2023). https://doi.org/10.1038/s12276-023-01119-5
- Noë, M., Mathios, D., Annapragada, A.V.et al. DNA methylation and gene expression as determinants of genome-wide cell-free DNA fragmentation. Nat Commun15, 6690 (2024). https://doi.org/10.1038/s41467-024-50850-8
! For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.




