Next-generation sequencing (NGS), also recognized as high-throughput sequencing, has evolved from PCR and Gene microarray technologies. This innovative sequencing method introduces reversible termination ends, allowing sequencing to occur concurrently with synthesis. The determination of DNA sequences is achieved by capturing newly added bases with specific markers during the DNA replication process. In a significant breakthrough, Roche introduced the first next-generation sequencer, the Roche 454, in 2005, marking the inception of the high-throughput sequencing era. And Illumina is becoming the most popular sequencing platform.
This sequencing technology holds paramount significance in life science and pharmaceutical research due to its capability to rapidly generate substantial data with a relatively short read length. In the NGS workflow, the initial step involves constructing the sample DNA, followed by machine testing and subsequent analysis. The construction of the sequencing library is a pivotal stage that dictates the success of NGS research. The quality of DNA and library construction products significantly influences parameters such as library conversion rate, sequencing depth, complexity, and homogeneity. Therefore, stringent quality control measures are imperative throughout the sequencing process.
What is Library Construction in NGS?
A DNA library encompasses a series of preparatory steps for DNA/RNA samples prior to sequencing. The original nucleic acid samples cannot be directly utilized for sequencing; only processed samples can meet the sequencing platform's requirements. These preparations involve tasks such as introducing essential end joints to DNA samples, a prerequisite for sequencing. In cases of insufficient sample volume, PCR amplification is employed to fulfill the machine's criteria. DNA library construction forms the foundation of next-generation sequencing library technology.
Quality Control in NGS Library Preparation Workflow may be a helpful article for the sequencing and analysis.
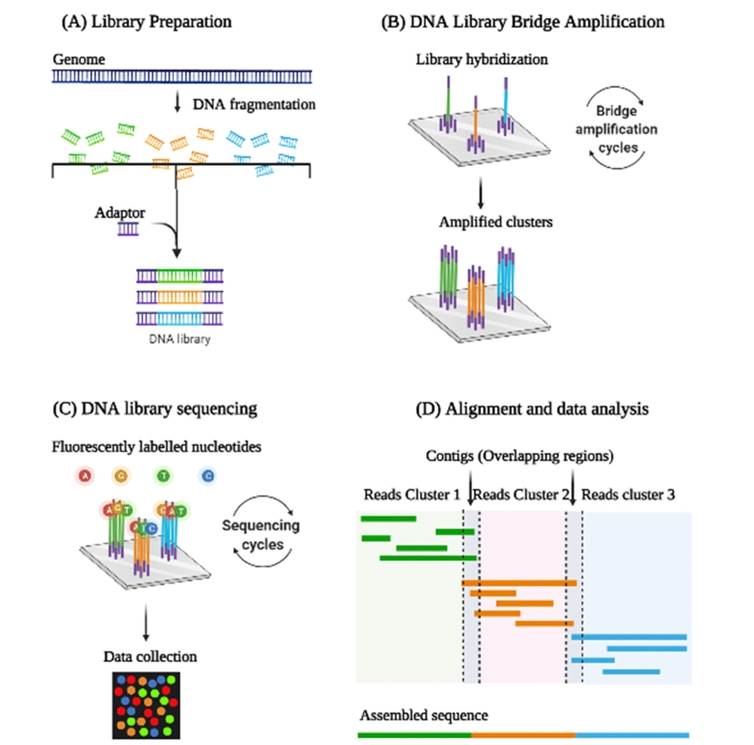 Represents the steps involved in Next Generation Sequencing: Library preparation and amplification, sequencing, and data analysis are the three important steps involved in NGS. (Selvakumar et al., 2022)
Represents the steps involved in Next Generation Sequencing: Library preparation and amplification, sequencing, and data analysis are the three important steps involved in NGS. (Selvakumar et al., 2022)
DNA library construction lies at the core of next-generation sequencing library technology. It involves the crucial step of adding end joints to the fragments under examination. Various methods exist for DNA library construction, categorized based on their approaches to joint formation:
- TA cloning junction construction
- Swift method
- Transposase library construction
- PCR amplicon construction
- Flat-end junction library construction
CD Genomics high-throughput sequencing and library construction services enable in-depth analysis of genomes, transcriptomes and epigenomes. Dive into the intricacies of library construction, a crucial step in this process, as it ensures the generation of high-quality data essential for robust genomic analysis.
Methods of NGS Library Preparation
TA Cloning Junction Library Construction
TA cloning junction library construction is presently the most prevalent method for library construction. The process entails the following steps:
- Genomic DNA fragmentation.
- End-repair of the fragmented DNA and addition of an A-tail.
- Attachment of the adapter.
- Sample concentration amplification through PCR amplification.
This technique necessitates the prior synthesis of target fragments with T-tailed adapters and A-tailed ends, subsequently linking them to fragmented samples via TA cloning facilitated by DNA ligase.
Swift Method
The Swift library construction method shares similarities with the TA cloning junction library construction method. It involves introducing P5 and P7 junction sequences at both ends of the fragment under examination. Following end repair, the P7 junction is initially connected at the 3' end, succeeded by the connection of the P5 junction at the 5' end. Subsequently, sample concentration is augmented through PCR amplification.
Transposase Library Construction
The cornerstone of transposase library construction lies in the Tn5 transposon, essentially a DNA fragment encoding the transposase gene. Traditional library construction entails DNA fragmentation, end repair, library amplification, and multi-step purification. However, leveraging Tn5 for library construction streamlines the process, condensing multiple steps into a single reaction.
The in vitro transposable elements of the Tn5 transposon utilized for library construction include the terminal sequence of the transposon, target DNA, transposase (Tnp), and Mg2+ (activator).
PCR Amplicon Library Construction
The amplicon library construction method employs a PCR reaction to introduce junctions at the ends of the target fragments, necessitating only two rounds of PCR and purification to yield the desired library. Initially, primers containing the universal sequence are paired with the target region. Subsequently, the sequencing junctions are linked through a PCR reaction in the second step.
Flat-End Junction Library Construction
The flat-end ligated adapter method entails attaching specific adapters to the ends of fragmented DNA fragments. This approach follows a series of steps including DNA sample fragmentation, end repair, adapter ligation, selective recovery of DNA fragments, PCR enrichment of the library, and purification of the PCR product. The sequencing process for constructing libraries with flat-end ligated adapters involves transient decreases in pH within the microenvironment, which are detected and recorded by a pH electrode to facilitate data reads.
Illumina DNA Library Construction
Please read our article: Illumina Next-Generation Sequencing (NGS): Principles and Workflow for more information.
Fragmentation
The initial phase of library construction involves addressing the read length limitations of sequencing machines. The extracted DNA cannot be directly sequenced due to these limitations. Therefore, various methods, such as ultrasonic fragmentation and enzyme digestion, are employed to precisely cut the DNA into fragments of suitable lengths. While mechanical methods may incur higher sample loss and complexity, enzymatic methods, particularly those utilizing the Tn5 transposase, are preferred for their cost-effectiveness and simplicity. The Tn5 transposon, originally identified in E. coli, comprises key components including IS50 sequences, Outside End (OE) sequences, Inside End (IE) sequences, and drug resistance genes.
End Repair/Addition of A-tail
Following fragmentation, DNA ends may exhibit flat or uneven characteristics. In this step, the ends of the DNA fragments are modified, and a distinctive A base is added to create a sticky end for subsequent junction primer attachment.
DNA fragments generated in the previous step, featuring 5'/3' sticky ends or flat ends, undergo end repair to convert all sticky ends to flat ends. For TA-joining, phosphorylation at the 5' end and the addition of an "A" at the 3' end are essential for complementary pairing with junctions possessing "T" sticky ends. This process involves the collaborative action of T4 DNA polymerase, T4 polynucleotide kinase, and Taq DNA polymerase.
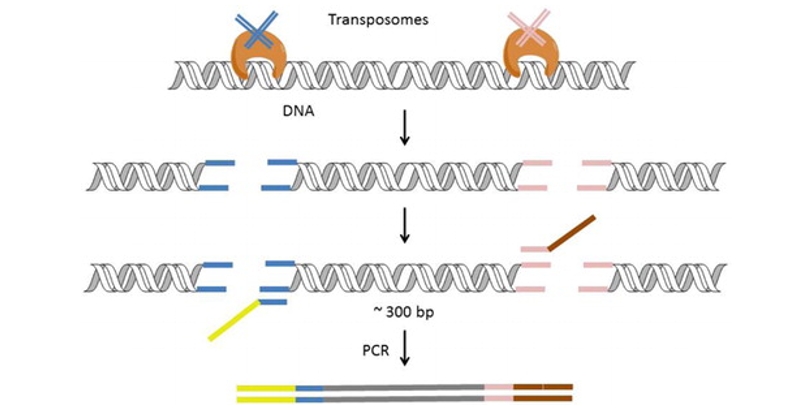 DNA library preparation using a transposase-based method (Nextera) developed by Illumina. (Head et al., 2014)
DNA library preparation using a transposase-based method (Nextera) developed by Illumina. (Head et al., 2014)
- T4 DNA polymerase exhibits 5'→3' DNA polymerase activity, synthesizing DNA in the 5'→3' direction and flattening 5' protruding ends. Additionally, its 3'→5' exonuclease activity flattens 3' protruding ends, transforming DNA fragments with sticky ends into flat-ended DNA.
- T4 Polynucleotide Kinase is essential for catalyzing the transfer of the γ-phosphate group of ATP to the hydroxyl end of the 5' end of the oligonucleotide strand, preparing the junctions for the subsequent joining step.
- Taq DNA Polymerase exhibits 5'→3' polymerase activity to synthesize DNA in the 5'→3' direction and deoxynucleotidyltransferase activity to add a nucleotide "A" to the 3' end of the PCR product. These intricate processes ensure the preparation of DNA fragments with optimized ends for the subsequent stages of Illumina DNA library construction.
Junctions
DNA fragments with appended A-tails exhibit prominent A-termini, facilitating their complementary pairing with junctions harboring T-termini. The primary objective of incorporating junctions is to append library tags and oligonucleotide sequences that complement the sequencing platform onto the ends of fragmented DNA.
Junctions play a pivotal role in the library, with Illumina platform Y-type junctions being widely utilized in sequencing. These junctions encompass P5/P7, Index, and Rd1/Rd2 SP sequences. P5/P7 sequences pair with the sequences on the sequencing chip, anchoring the fragments for testing on the Flowcell to complete bridge amplification. The Index distinguishes between different samples in the onboard sequencing mixed library, and Rd1/Rd2 SP serves as the primer binding region for Read1 and Read2 sequencing. Junction ligation typically involves T4 DNA Ligase, repairing single-stranded cuts in double-stranded DNA and reconnecting adjacent nucleotides. In junction ligation, junctions with "T" sticky ends and "A" sticky ends can be seamlessly combined, forming a complete double strand.
PCR Amplification
Due to the previously added junction, direct amplification is achieved using primers complementary to the junction. The non-complementary ends of the previously added "Y" junctions necessitate an intermediary step before direct sequencing. To sequence multiple samples concurrently, indexes/barcodes can be added to differentiate between different samples. This step not only aids in distinguishing various libraries during subsequent sample analysis but also introduces oligonucleotide sequences complementary to the sequencer at both ends through PCR, specifically P5/P7.
4 Milestones in DNA Sequencing Technology
First Breakthrough: Direct Reading
- Pre-Direct Reading: The original sequencing method predating direct reading involved techniques such as molecular cloning, PAGE, and radiographic autoradiography.
- SBC Method: A chemical degradation reaction using specific reagents to directly degrade DNA molecules. Gilbert received the Nobel Prize in Chemistry in 1980 for inventing the SBC method.
- SBS Method: Based on enzymatic synthesis reaction and DNA synthesis, also known as the Sanger method or double deoxyribonucleic acid terminal termination method. This method offers advantages such as non-toxic reagents, ease of operation, stable results, high accuracy, and excellent reproducibility.
Second Breakthrough: Automation
- Pioneers of Automated Sequencing: Akiyoshi Wada, Wilhelm Ansorge.
- The key breakthrough in automating the SBS method came with Leroy Hood's invention of the automated four-color fluorescent-labeled SBS sequencer. This innovation played a crucial role in supporting and launching the Human Genome Project (HGP).
Third Breakthrough: Scale-Up
- The emergence and enhancement of capillary electrophoresis sequencing technology facilitated scale-up.
- In 1998, ABI introduced the ABIPrism 3700 capillary sequencer (ABI3700), enabling direct scaling up of sequencing technology. The subsequent introduction of the MegaBACE series of sequencers significantly contributed to the early completion of the HGP.
Fourth Breakthrough: Massively Parallel Sequencing
This represents a substantial leap forward, characterized by the ability to sequence DNA in a massively parallel manner, resulting in a rapid and simultaneous analysis of multiple fragments. One of the key features of sequencing is its significant contribution to the precipitous drop in sequencing costs.
FAQ: Create A Sequencing Library
1. What are the primary steps involved in DNA library construction?
The main parts of DNA library construction involve:
- Fragmentation and End Repair: Breaking down the DNA into manageable fragments and repairing the ends to ensure proper ligation.
- Ligation: Joining the repaired DNA fragments to adapters or vectors for sequencing.
- PCR (Polymerase Chain Reaction): Amplifying the ligated DNA to increase the quantity for sequencing.
Note: Bead purification steps are omitted here.
Additionally, constructing a library from RNA samples involves an extra step due to the nature of RNA. It requires reverse transcription of RNA into complementary DNA (cDNA) before proceeding with the above library construction process. However, the fundamental principle remains the same.
2. Why is it necessary to fragment gDNA samples prior to NGS library preparation?
Illumina sequencers typically read fragments within the range of 50-600 base pairs, although this range can vary depending on the sequencing chips and reagents used. Conversely, most intact human genomic DNA (gDNA) samples exceed 10 kilobases (kb) in length. Therefore, breaking down these large fragments into smaller pieces is essential for successful library construction. This fragmentation prerequisite holds true for the MGI platform as well.
Table 1 Recommended Sequencing Read Lengths for Different Applications on Illumina Sequencing Platforms
3. Why is it essential to incorporate an adapter following DNA fragmentation? What significance does it hold?
A complete adapter comprises three components: universal primers (P5&P7), index (i7/i5), and sequencing primers (SP1&SP2).
- Universal primers facilitate cluster generation, while sequencing primers are essential for sequencing the insert.
- The splice sequence is determined by the sequencing platform.
- Indexing serves to differentiate various samples within the same batch of sequencing data. When sequencing multiple samples on the same chip (≥2), indexing is crucial for sample discrimination. However, if only one sample occupies a flow cell, indexing becomes unnecessary, although universal primers (P5&P7) and sequencing primers (SP1&SP2) remain essential.
Note: While there are multiple methods to add a complete junction to a sample, the resulting library's junction sequence remains consistent. We intend to dedicate an article to elucidate the diverse structures and connections of junctions in the future.
4. Why do PCR and PCR-free libraries exist, and which is more prevalent?
PCR serves as a method to boost sample volume. When the initial input volume is insufficient, PCR replication is necessary to attain the requisite library volume for sequencing. Conversely, ample starting volume allows for completing junction ligation, purification, and subsequent sequencing without PCR amplification.
However, PCR libraries are more widespread due to several reasons:
- Insufficient Starting Volume: Most samples lack the initial volume required for direct onboarding.
- Specialized Physical Structure: Libraries not amplified by PCR possess a unique Y-shaped junction, which can introduce biases in library quality control assessments.
- Residual Junctions: The high ratio of junctions to templates and inadequate purification result in significant residual junctions, diminishing overall library quality.
5. Why do various kits impose distinct requirements for sample starting volumes?
The starting volume capability of a kit primarily hinges on the sensitivity, specificity, and stability of its respective enzymes. Kits catering to lower starting volumes (e.g., nasopharyngeal swab samples for COVID-19 testing) demand heightened enzyme efficacy, consequently reflecting in higher prices. Additionally, kits tailored for low starting volume samples often boast proprietary patents and distinct advantages, further contributing to their elevated costs. Please contact our technical team for more information.
References:
-
Selvakumar, Sushmaa Chandralekha, et al. "CRISPR/Cas9 and next generation sequencing in the personalized treatment of Cancer." Molecular Cancer 21.1 (2022): 83.
- Head, Steven R., et al. "Library construction for next-generation sequencing: overviews and challenges." Biotechniques 56.2 (2014): 61-77.

 Sample Submission Guidelines
Sample Submission Guidelines


