
 Sample Submission Guidelines
Sample Submission Guidelines
What is De Novo Whole Genome Sequencing
Whole-genome de novo sequencing, also known as de novo sequencing, enables the sequencing of a species without relying on any pre-existing genetic sequence information. Through bioinformatic methods, the obtained sequences are assembled and aligned, resulting in a detailed genomic map of the species under study. This approach facilitates the acquisition of entire genomic sequences for a diverse array of organisms, including animals, plants, bacteria, and fungi, thereby propelling forward our understanding and research of these species.
Upon finalizing the whole-genome sequencing process, a comprehensive genomic database can be established for the species in question. This database serves as a robust platform for subsequent post-genomic studies, enhancing the capability to undertake gene mining and functional validation work effectively. The advent and application of next-generation high-throughput sequencing technologies have rendered it feasible to procure the genomic sequences of various organisms in a manner that is both cost-effective and efficient. Consequently, these advancements are significantly driving research and expanding our knowledge base across the biological sciences, encompassing animals, plants, bacteria, and fungi.
Overall, employing whole-genome de novo sequencing techniques enables the acquisition of complete genomic sequences of various organisms, such as plants and animals, thus stimulating subsequent research endeavors pertaining to these species. Upon completion of the whole-genome sequencing, a comprehensive genomic database can be established for the species in question. This database serves as an efficient platform for post-genomic studies, facilitating gene discovery and functional verification by providing critical DNA sequence information.
Our De Novo Whole Genome Sequencing Service
CD Genomic offers a one-stop solution for de novo genome sequencing services, covering experimental design, sample preparation, sequencing, and bioinformatics analysis, aiming to provide genomic solutions for your research. Our de novo genome sequencing services include:
- De novo sequencing of animal and plant genomes.
- De novo sequencing of bacterial genomes.
- De novo sequencing of fungal genomes.
- De novo sequencing of metagenomes.
Advantages of Our De Novo Whole Genome Sequencing Service
- Platform Advantages: Obtaining high-quality genomic maps
- High-depth sequencing, High accuracy
- Rich data analysis content: Standard analysis, advanced analysis, customized analysis
- Extensive experience in genome sequencing and analysis: Successfully completed numerous large-scale genome sequencing projects
- Customized genome sequencing solutions: Personalized genome sequencing solutions can be tailored according to specific requirements.
Application of De Novo Whole Genome Sequencing
- Obtaining the reference sequence of a species
- Studying the origin and evolutionary history of a species
- Mining functional genes
- Establishing a species database
- Research on the genome of a new species
- Analysis of complex genome structures
- Population genetics and genomic diversity
- Disease genomics
- Functional genomics and regulatory elements
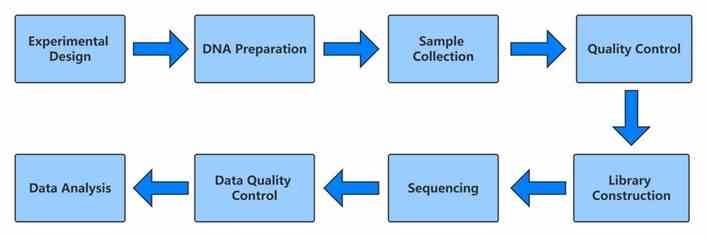
Workflow of De Novo Whole Genome Sequencing
Our De Novo Whole Genome Sequencing Service starts with DNA extraction, followed by state-of-the-art sequencing and rigorous bioinformatics analysis. We provide accurate genome assembly and annotation to uncover genetic insights effectively.
Service Specifications
| Sample Requirements Whole Genome Sequencing:
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis
|
Analysis Pipeline
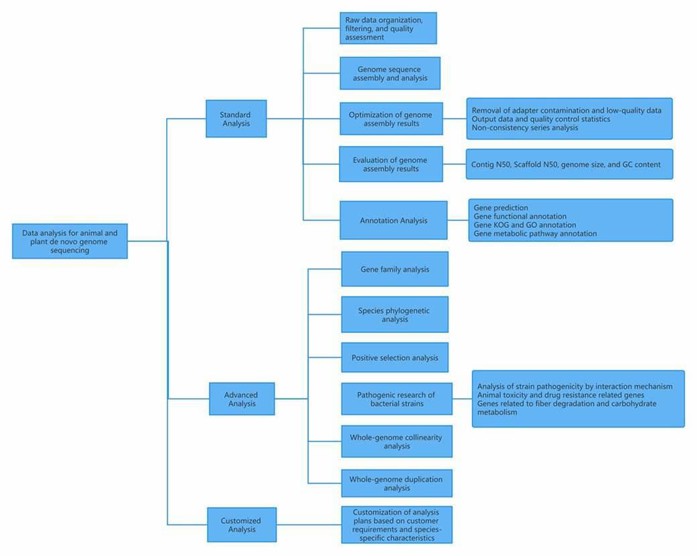
I. Data analysis for animal and plant de novo genome sequencing
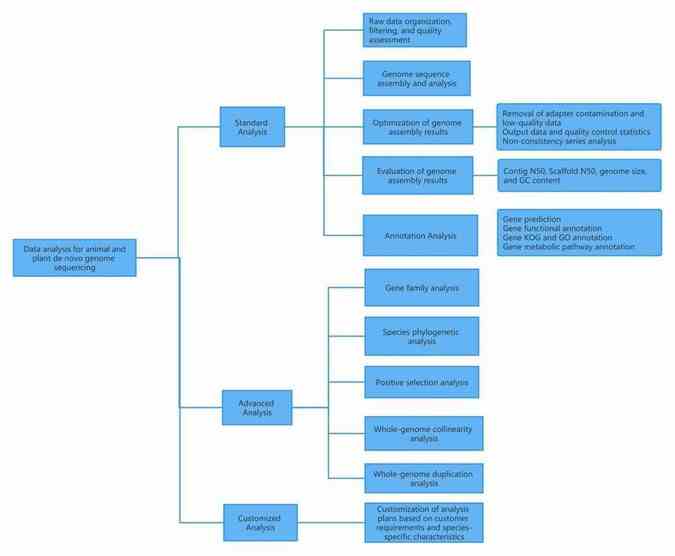
II. Data analysis for bacterial and fungal de novo genome sequencing
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in De Novo Whole Genome Sequencing for your writing (customization)
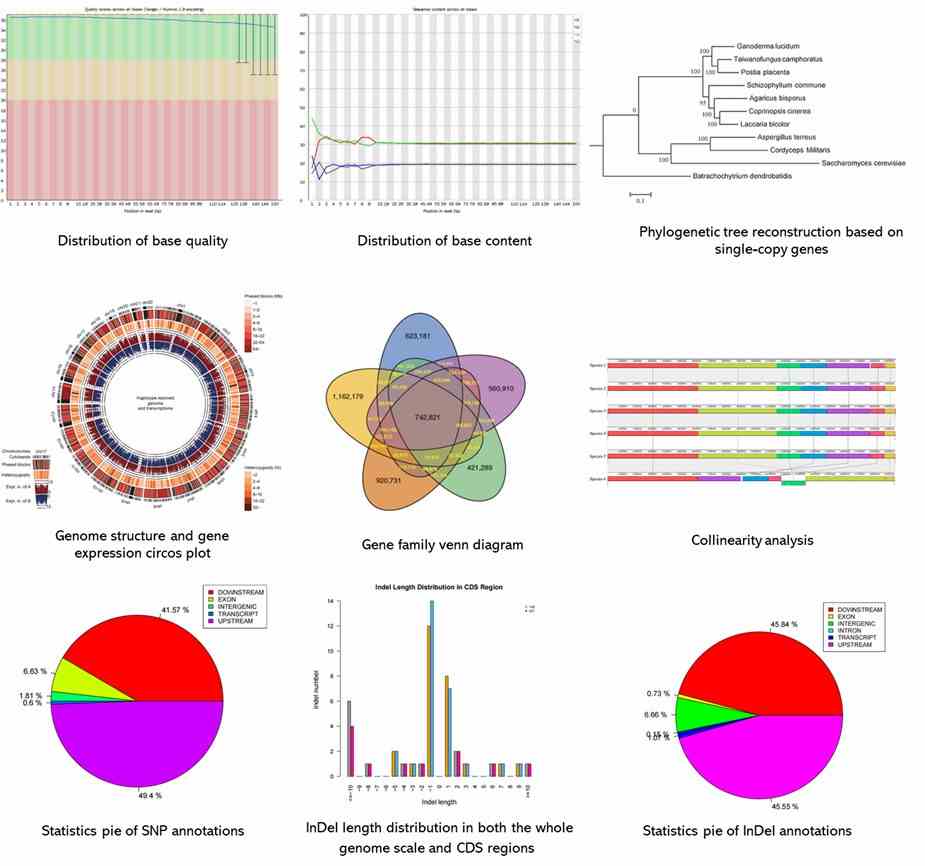
Partial results are shown below:
1. What is the difference between de novo and reference assembly?
De novo assembly involves constructing a genome sequence from scratch without relying on a pre-existing reference genome, which is particularly useful for species without a known genome sequence. Reference assembly, on the other hand, uses an existing genome sequence as a template to align and map sequencing reads, allowing for the identification of genetic variations in the studied genome relative to the reference.
2. How to assess the quality of genome de novo assembly?
The quality of genome de novo assembly is commonly evaluated using metrics such as contig N50 and scaffold N50. The N50 value is determined by arranging the assembled contigs or scaffolds from largest to smallest and identifying the length of the contig or scaffold at the point where the cumulative length surpasses 50% of the total assembly length. This value provides significant insight into the continuity and completeness of the assembled sequences. Additional measures like N70 and N90 are calculated similarly, with the percentage thresholds adjusted to 70% and 90%, respectively. These metrics collectively serve to comprehensively evaluate the integrity and reliability of the genome assembly.
3. Is the DNA used in survey sequencing and whole-genome de novo sequencing required to be the same?
In principle, the DNA used for both survey sequencing and de novo sequencing should originate from the same individual. If the amount of DNA is insufficient for the entire de novo sequencing project, it is recommended that the DNA for the short-read library comes from the same individual. For third-generation sequencing long-read or even ultra-long-read libraries, DNA from another individual within the same population may be used.
Comparison of Arachis monticola with Diploid and Cultivated Tetraploid Genomes Reveals Asymmetric Subgenome Evolution and Improvement of Peanut
Journal: Advanced Science
Impact factor: 15.804
Published: 28 November 2019
Background
Peanut is a vital global oilseed legume, originating from South America and cultivated predominantly in Asia and Africa. Its evolution from wild diploid species to cultivated tetraploid forms offers insights into polyploid genome evolution and crop domestication, crucial for enhancing global food and oil security. The authors utilized genome sequencing and comprehensive genomic analysis to study the evolution and domestication of peanuts.
Materials & Methods
Sample Preparation
- Peanut plants
- Fresh leaves
- DNA extraction
- RNA extraction
- Repetitive elements (TE) Annotation
- Gene Prediction and Annotation
- lncRNA Identification
- Population SNPs Detection
- Population Phylogenetic Relationship Construction
- Principal Component Analysis
Results
The study resequenced genomes of 17 wild diploids and 30 wild/cultivated tetraploids, performing phylogenetic and PCA analyses to classify them into wild and cultivated groups. It showed that cultivated tetraploids evolved from wild tetraploids. A whole genome duplication occurred ≈58 million years ago, accelerating sequence divergence in tetraploid peanuts. The study reconstructed the evolution of allotetraploid peanuts, highlighting hybridization and domestication events around 4500 years ago, impacting agronomic traits and resistance characteristics.
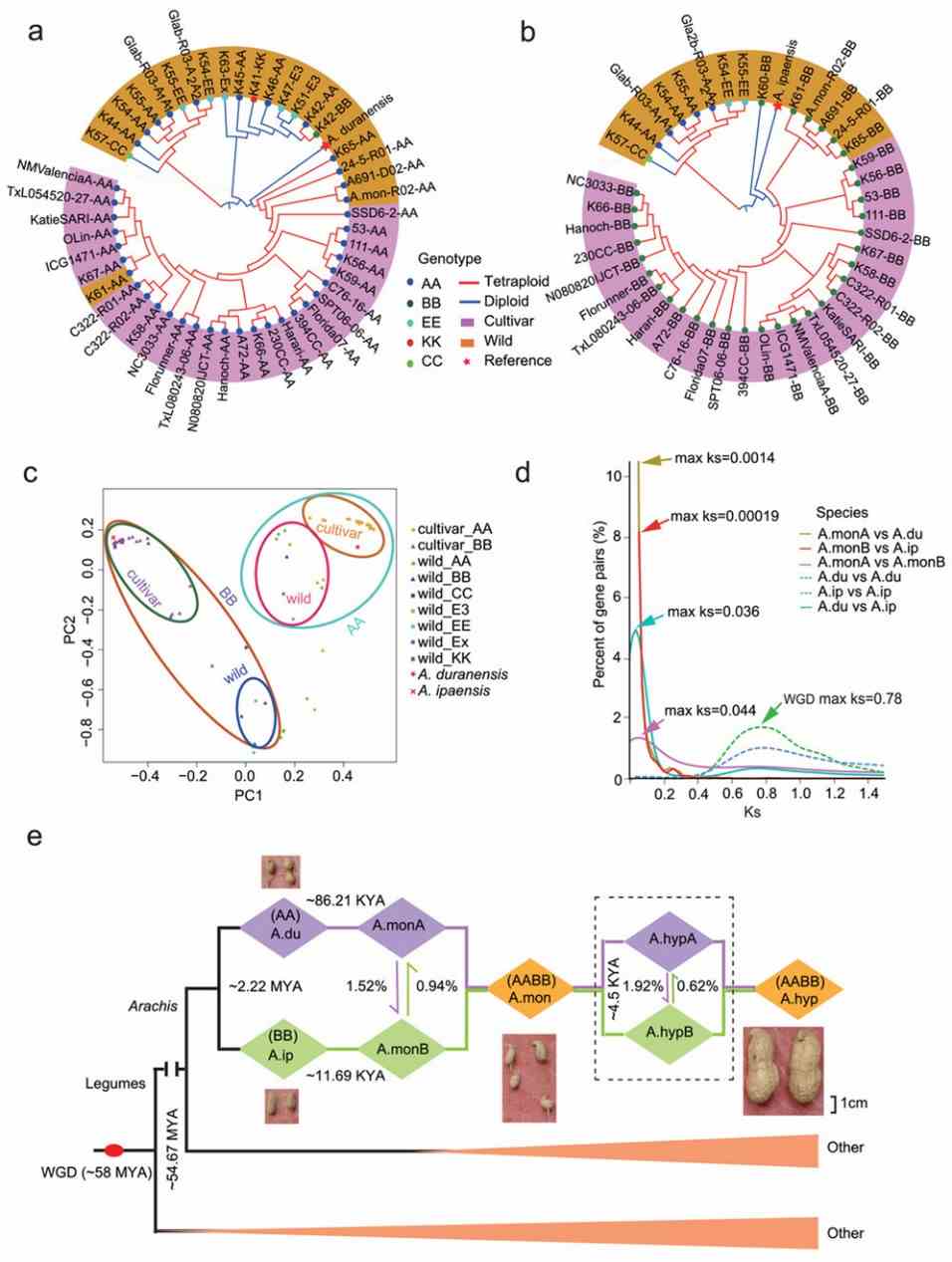 Fig 1. Subgenome origins and phylogenetic analysis of wild and cultivated peanut lines.
Fig 1. Subgenome origins and phylogenetic analysis of wild and cultivated peanut lines.
In tetraploid peanuts, asymmetric evolution between subgenomes was evident. Sequence exchanges favored A to B subgenomes, influencing gene family sizes and pathways like flavonoid biosynthesis. Expression bias during pod development indicated divergent roles of A and B subgenomes. Selection pressures differed between wild and cultivated forms, highlighting asymmetric impacts on gene evolution and expression in peanuts.
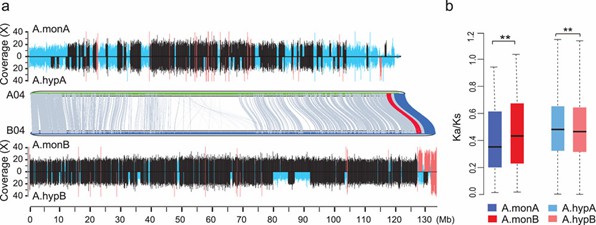 Fig 2. Asymmetric subgenome evolution of allotetraploid peanuts.
Fig 2. Asymmetric subgenome evolution of allotetraploid peanuts.
SVs (deletions and insertions) influence gene expression and traits in peanuts.Analysis of homoeologous pairs reveals different selection pressures on SV genes between subgenomes during peanut domestication, indicating distinct patterns of SV accumulation and evolutionary impacts.
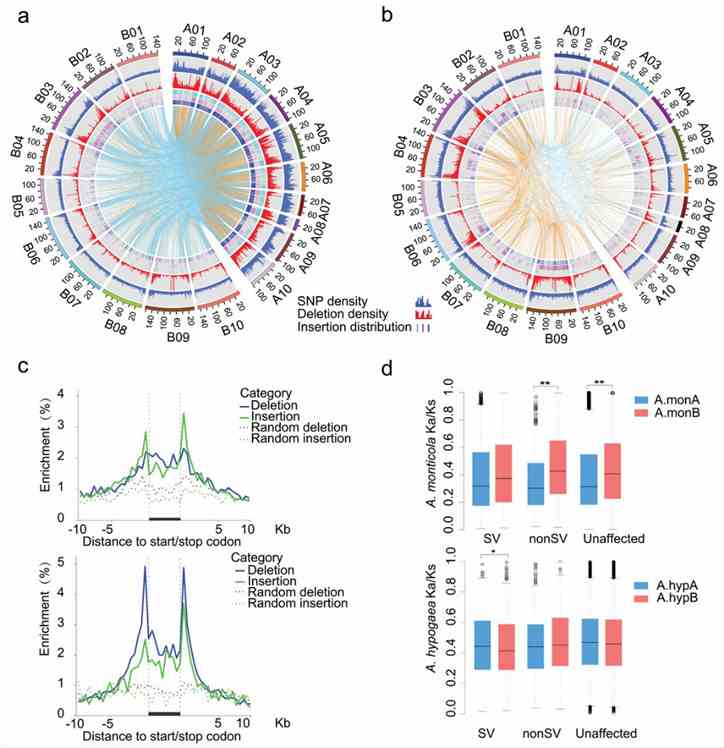 Fig 3. Asymmetric SV accumulation from wild diploids to allotetraploid peanuts.
Fig 3. Asymmetric SV accumulation from wild diploids to allotetraploid peanuts.
Conclusion
The high-quality sequence of wild tetraploid peanut bridges the genomic gap between diploid and cultivated species, revealing asymmetric evolution in subgenomes and providing valuable resources for studying polyploid evolution and peanut improvement.
Reference
- Yin D, Ji C, Song Q, Zhang W, Zhang X, Zhao K, Chen CY, Wang C, He G, Liang Z, Ma X. Comparison of Arachis monticola with diploid and cultivated tetraploid genomes reveals asymmetric subgenome evolution and improvement of peanut. Advanced Science. 2020, 7(4):1901672.
Here are some publications that have been successfully published using our services or other related services:
Distinct functions of wild-type and R273H mutant Δ133p53α differentially regulate glioblastoma aggressiveness and therapy-induced senescence
Journal: Cell Death & Disease
Year: 2024
High-Density Mapping and Candidate Gene Analysis of Pl18 and Pl20 in Sunflower by Whole-Genome Resequencing
Journal: International Journal of Molecular Sciences
Year: 2020
Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI
Journal: New phytologist
Year: 2017
Generation of a highly attenuated strain of Pseudomonas aeruginosa for commercial production of alginate
Journal: Microbial Biotechnology
Year: 2019
Combinations of Bacteriophage Are Efficacious against Multidrug-Resistant Pseudomonas aeruginosa and Enhance Sensitivity to Carbapenem Antibiotics
Journal: Viruses
Year: 2024
Genome Analysis and Replication Studies of the African Green Monkey Simian Foamy Virus Serotype 3 Strain FV2014
Journal: Viruses
Year: 2020
See more articles published by our clients.


