Advancements in genomic technologies have significantly improved our understanding of gene expression and the complexity of RNA transcripts. As these technologies evolve, new possibilities emerge for studying RNA molecules, their structures, functions, and regulatory mechanisms, all of which are critical for advancing biological research and its broad array of applications. To address these challenges, various sequencing technologies have been developed, each offering distinct advantages suited to particular research needs. Among these, Iso-Seq stands out as a groundbreaking innovation. This method, which directly captures full-length RNA transcripts, overcomes the traditional difficulties associated with computational assembly, providing researchers with a more accurate and comprehensive view of transcriptomic data. Developed by Pacific Biosciences, Iso-Seq employs SMRT sequencing technology, producing long, high-quality reads that enable a deeper exploration of RNA diversity. This paper explores the principles behind Iso-Seq, its benefits, applications, and compares its performance with other sequencing technologies, emphasizing Iso-Seq's strengths in addressing specific genomic challenges.
Overview of Iso-Seq
Iso-Seq represents a significant advancement in sequencing technology by facilitating the direct sequencing of full-length RNA transcripts, generating high-quality, long reads. Traditional sequencing approaches, especially short-read RNA sequencing, struggle to fully capture large or complex RNA transcripts. These methods typically depend on computational assembly of fragmented sequences, which can introduce errors, particularly when dealing with longer transcripts. Iso-Seq resolves these issues by sequencing complete complementary DNA (cDNA) molecules in a continuous manner, thereby offering a more accurate and complete representation of gene expression and transcript diversity. This direct sequencing approach is especially advantageous for applications such as gene discovery, transcript annotation, and the study of alternative splicing events—each of which requires precise and comprehensive transcript representation, a requirement that Iso-Seq effectively fulfills.
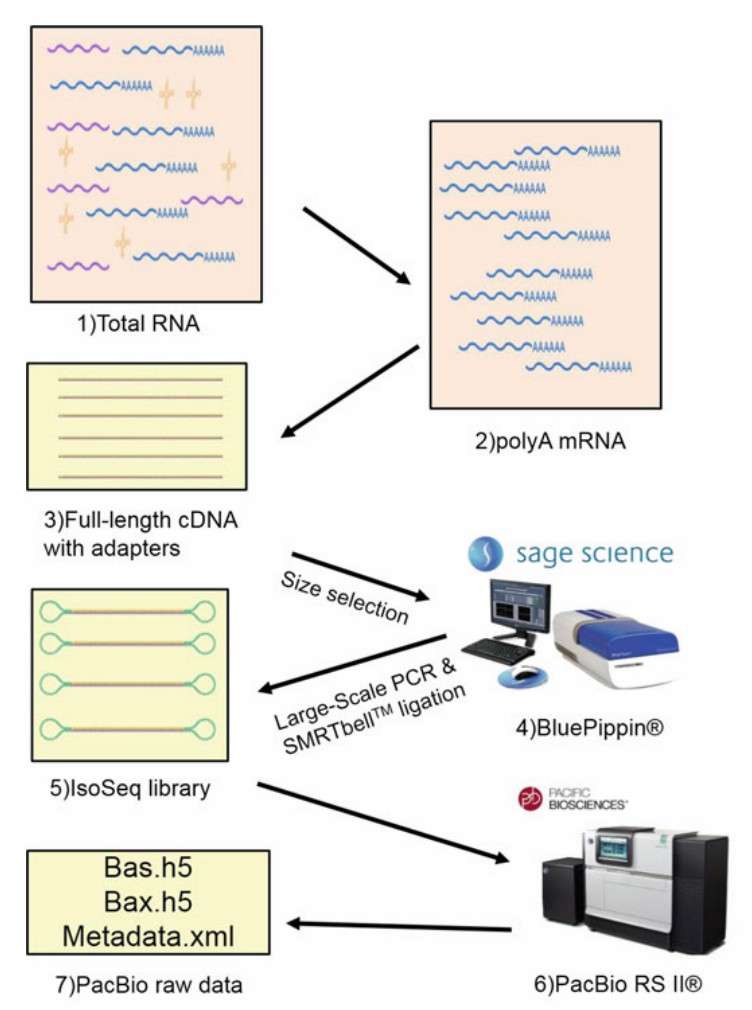 Overview of the Iso-Seq protocol (Gonzalez-Garay, 2016)
Overview of the Iso-Seq protocol (Gonzalez-Garay, 2016)
Principles and Advantages of Iso-Seq
The primary strength of Iso-Seq lies in its ability to generate extraordinarily long reads, averaging around 10 kilobases (kb). This feature enables the sequencing of entire RNA molecules, including the often-overlooked untranslated regions (UTRs) at both the 5' and 3' ends. UTRs play a critical role in gene regulation, and their inclusion in the sequencing process ensures that researchers capture the complete picture of transcript isoforms. In contrast to other sequencing methods that require complicated assembly processes, Iso-Seq directly sequences full-length transcripts, minimizing the risk of errors related to misassembly. This direct approach not only enhances the accuracy of transcriptomic data but also facilitates the identification of intricate transcript features, such as alternative splicing, intron retention, and other regulatory mechanisms that are essential for understanding gene expression regulation. Iso-Seq's capacity to produce long, high-quality reads makes it particularly suitable for capturing the full complexity of gene expression and transcript variability.
Applications of Iso-Seq
Iso-Seq serves as an invaluable tool for a range of applications, particularly in gene discovery and transcript annotation. By enabling the identification of previously uncharacterized transcripts and rectifying inaccuracies in existing gene models, Iso-Seq helps researchers generate a more complete and accurate transcriptome map. Furthermore, Iso-Seq is crucial for studying alternative splicing, a key regulatory process that allows a single gene to produce multiple RNA isoforms. This process is central to gene expression regulation and the diversification of protein functions. Iso-Seq's ability to capture full-length RNA molecules is indispensable for uncovering the molecular mechanisms underlying these processes. Additionally, the technology's capacity to provide a comprehensive view of gene expression is vital for investigating complex regulatory networks that control cellular functions and biological processes. Iso-Seq's versatility makes it a powerful tool for various biological studies, ranging from basic gene function research to the investigation of intricate regulatory pathways.
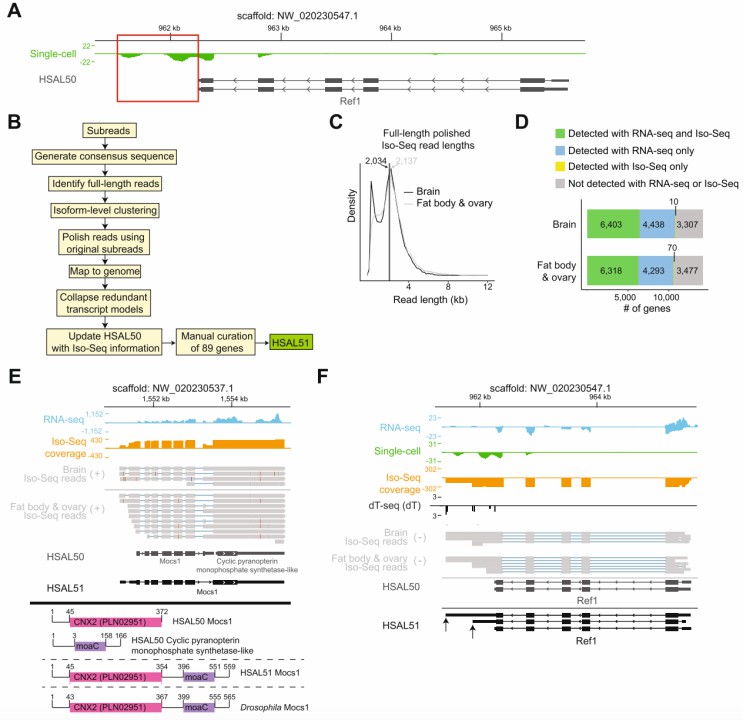 Iso-Seq improves gene models in Harpegnathos (Shields et al., 2021)
Iso-Seq improves gene models in Harpegnathos (Shields et al., 2021)
Services you may interested in
Want to know more about the details of Iso-seq? Check out these articles:
Comparison of Iso-Seq with Other Sequencing Technologies
Although Iso-Seq offers many advantages, it is important to evaluate it in the context of other sequencing technologies, such as traditional RNA-Seq and other long-read sequencing platforms, like Oxford Nanopore. Each of these technologies has unique strengths, weaknesses, and suitability for specific genomic research. This section aims to compare Iso-Seq with traditional RNA-Seq, other long-read technologies, and hybrid approaches that integrate multiple sequencing methods. The objective is to highlight Iso-Seq's strengths and limitations in relation to other available platforms.
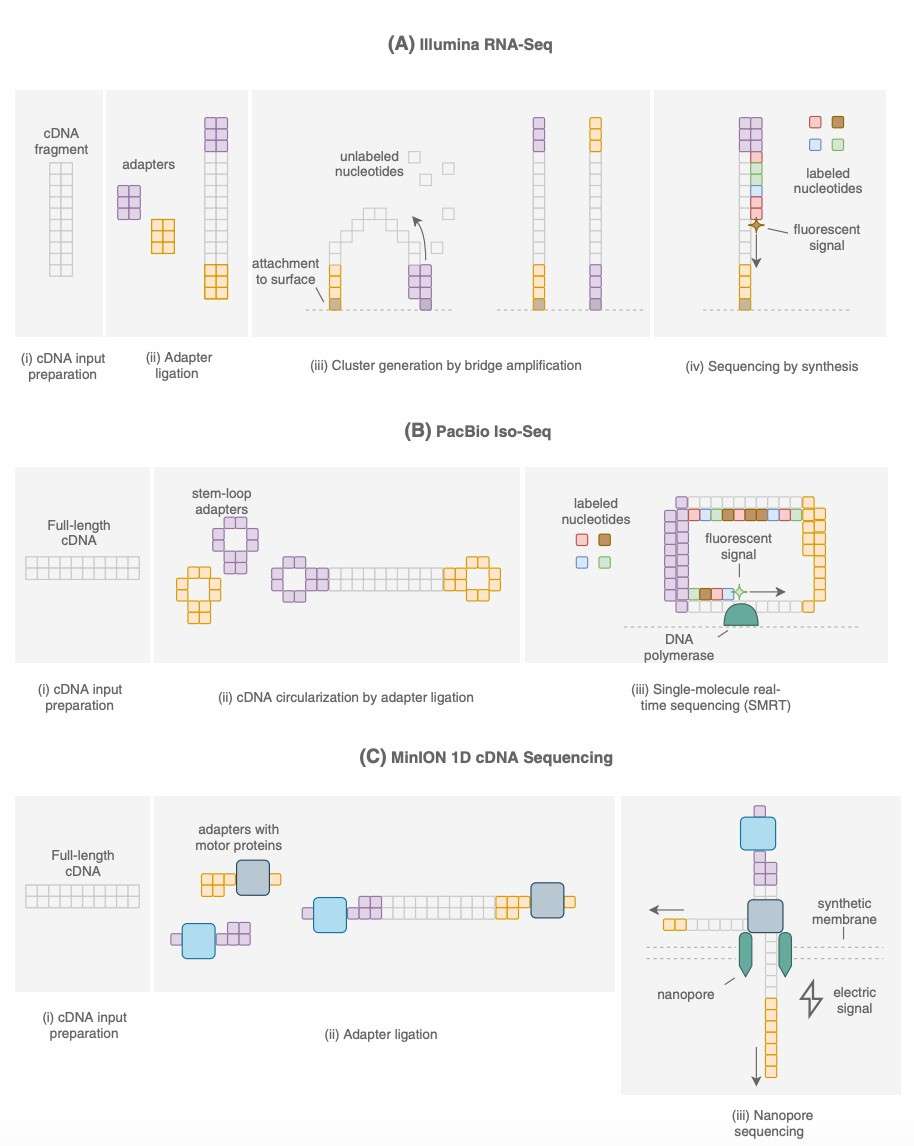 Comparison of the Various Sequencing Platforms (Boldogkői et al., 2019)
Comparison of the Various Sequencing Platforms (Boldogkői et al., 2019)
Traditional RNA-Seq
Traditional RNA-Seq, a widely utilized short-read sequencing method, has significantly advanced transcriptomics. However, it is constrained by its relatively short read lengths of 100–150 base pairs, making it difficult to capture long RNA transcripts or complex splicing patterns. As a result, RNA-Seq often necessitates computational assembly to reconstruct fragmented sequences, leading to incomplete or inaccurate transcript representations, particularly for genes with multiple isoforms or those undergoing alternative splicing. In contrast, Iso-Seq directly sequences full-length RNA transcripts, eliminating the need for assembly and providing a more complete and accurate depiction of the transcriptome. While RNA-Seq remains a cost-effective method for large-scale gene expression profiling, it faces challenges in detecting full-length transcript isoforms. Combining RNA-Seq with Iso-Seq offers an enhanced strategy, leveraging RNA-Seq's high throughput and Iso-Seq's ability to capture full-length transcripts, resulting in a more comprehensive transcriptomic analysis.
Other Long-Read Technologies (e.g., Oxford Nanopore)
Other long-read sequencing platforms, such as Oxford Nanopore, also enable the direct sequencing of full-length RNA molecules. However, there are notable differences in performance between these technologies. Oxford Nanopore generally generates shorter reads compared to Iso-Seq, with an average length of around 2-3 kilobases. Moreover, Oxford Nanopore reads tend to have lower accuracy compared to Iso-Seq, which is renowned for producing high-quality long reads of approximately 10 kb. Despite these differences, Oxford Nanopore offers advantages such as portability, ease of use, and lower operational costs, making it an attractive choice for certain applications. However, the reduced accuracy and shorter read lengths of Oxford Nanopore can limit the reliability of transcriptomic analyses, especially when analyzing complex genomic regions or performing detailed transcript annotation. While Iso-Seq delivers superior read quality and length, it comes at a higher cost, which might be prohibitive for large-scale sequencing projects. Researchers need to consider factors such as read length, accuracy, cost, and scalability when choosing between Iso-Seq and other technologies like Oxford Nanopore.
Hybrid Approaches (Combining Iso-Seq with Other Technologies)
Hybrid sequencing approaches that combine Iso-Seq with other technologies, such as short-read RNA-Seq or alternative long-read platforms, provide a powerful and comprehensive solution for transcriptomic analysis. By integrating Iso-Seq's high-quality, full-length reads with RNA-Seq's high-depth coverage, researchers can capitalize on the strengths of both technologies. This combination enhances the overall accuracy and completeness of transcriptomic studies, providing both detailed, full-length data and the necessary throughput for large-scale projects. However, hybrid approaches often involve more complex bioinformatics workflows to merge and analyze the data effectively. These methods are particularly useful in areas such as plant genomics, where the complexity of the transcriptome requires the use of multiple sequencing technologies to gain a more thorough understanding of gene expression and regulation. Researchers must possess both technical expertise and computational resources to successfully implement these hybrid strategies.
Comparative Analysis of Key Features of Iso-Seq and Other Technologies
When selecting a sequencing platform, researchers must carefully evaluate several key factors, including read length, accuracy, cost, scalability, and data complexity. These aspects directly influence the suitability of a technology for specific research goals. The following comparative analysis focuses on these critical features, offering guidance for researchers in choosing the most appropriate sequencing platform based on their needs.
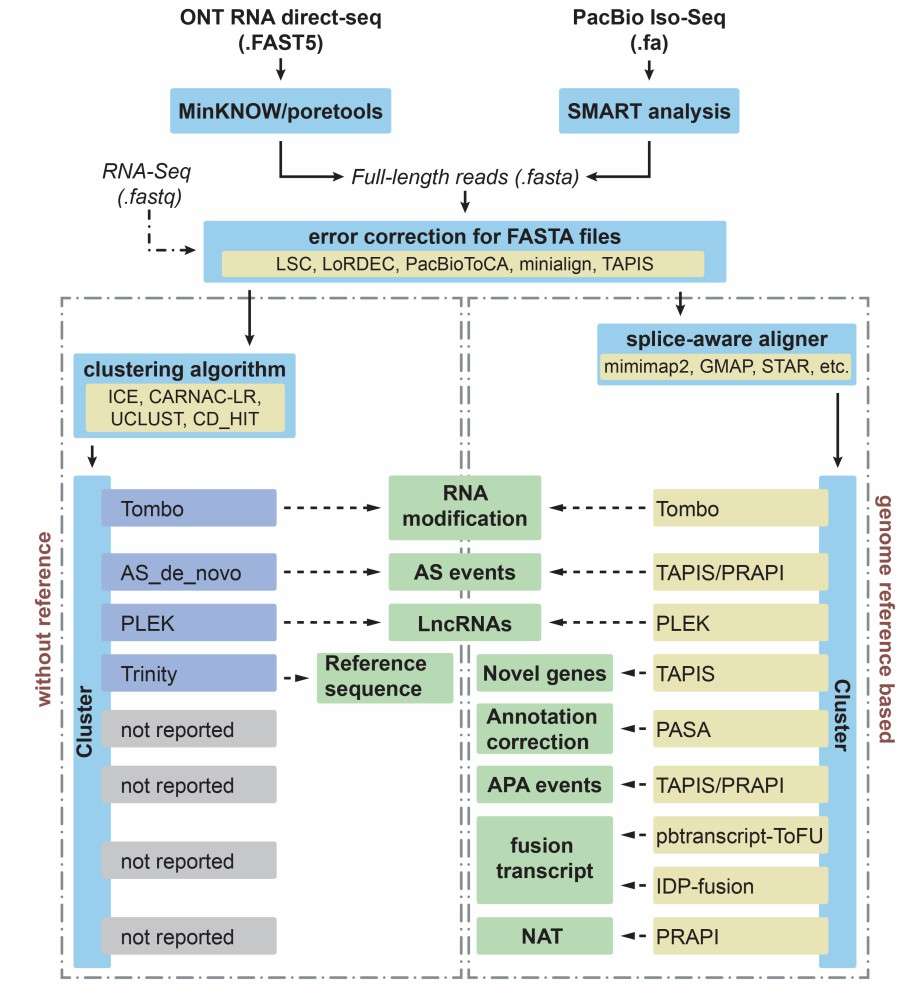 Different applications and bioinformatics solutions for PacBio Iso-Seq and Nanopore direct RNA sequencing in plants (Zhao et al., 2019)
Different applications and bioinformatics solutions for PacBio Iso-Seq and Nanopore direct RNA sequencing in plants (Zhao et al., 2019)
Read Length and Accuracy
One of Iso-Seq's most distinguishing features is its long read length. With an average read length of 10 kilobases, Iso-Seq can sequence entire RNA transcripts, including their UTRs, which are crucial for understanding gene regulation. In contrast, traditional RNA-Seq generates shorter reads, typically around 100–150 base pairs, which may not capture the full length of long transcripts or miss important transcript features such as UTRs. Iso-Seq's longer reads also provide superior accuracy, particularly for transcript assembly, since the technology does not require computational reconstruction of fragmented reads. The combination of long read lengths and high accuracy makes Iso-Seq an ideal choice for comprehensive studies of gene expression, alternative splicing, and transcript diversity.
Cost and Scalability
Despite its clear advantages, Iso-Seq is more expensive compared to traditional RNA-Seq, making it better suited for smaller-scale studies or projects focused on generating high-quality transcript data. In contrast, RNA-Seq is a more cost-effective option for large-scale gene expression studies, offering greater scalability. However, this cost efficiency comes at the cost of read length and transcript accuracy. Researchers must consider factors such as project scale, budget, and research objectives when choosing between Iso-Seq and RNA-Seq, as the higher cost of Iso-Seq may limit its applicability in larger studies.
Data Complexity and Analysis Requirements
Iso-Seq produces more complex data due to its longer read lengths and higher data quality. This complexity requires advanced bioinformatics tools for processing, alignment, and analysis, making the workflow more computationally intensive. However, the rich data generated by Iso-Seq can provide valuable insights into the transcriptome that may be difficult to obtain with other sequencing technologies. In contrast, short-read RNA-Seq generates simpler data that are easier to analyze but may miss important details related to complex transcript features such as alternative splicing. Researchers must weigh the trade-offs between detailed data and increased analysis complexity when selecting between sequencing technologies.
Applications and Suitability for Different Research Questions
Iso-Seq excels in applications that require a detailed understanding of the full-length transcriptome, such as gene discovery, alternative splicing analysis, and the identification of non-coding RNAs. Its ability to sequence entire transcripts without assembly makes it an essential tool for exploring gene structures and regulatory mechanisms. In contrast, traditional RNA-Seq is better suited for large-scale studies where cost and scalability are paramount. While RNA-Seq excels in gene expression profiling, it may miss important details related to transcript structure and diversity. Researchers must select the most suitable technology based on their specific research questions, balancing the trade-offs between data quality, cost, and scalability.
Conclusion
Iso-Seq has revolutionized transcriptomic analysis by offering a highly effective method for sequencing full-length RNA transcripts with exceptional accuracy and read length. It is particularly beneficial for studies focused on gene discovery, annotation, and the analysis of complex splicing events. However, its higher cost may limit its application in large-scale projects, making traditional RNA-Seq a more viable option in such cases. For more comprehensive studies, hybrid approaches that combine Iso-Seq with RNA-Seq or other technologies offer a promising solution, leveraging the strengths of each platform to provide a more complete understanding of the transcriptome. Ultimately, the choice of sequencing platform depends on the research objectives, available resources, and desired outcomes.
References:
- Boldogkői, Zsolt et al. "Long-Read Sequencing - A Powerful Tool in Viral Transcriptome Research." Trends in microbiology. 27,7 (2019): 578-592. doi:10.1016/j.tim.2019.01.010
- Gonzalez-Garay, Manuel L. "Introduction to isoform sequencing using pacific biosciences technology (Iso-Seq)." Transcriptomics and gene regulation (2016): 141-160. doi:10.1007/978-94-017-7450-5_6
- Shields, Emily J et al. "Genome annotation with long RNA reads reveals new patterns of gene expression and improves single-cell analyses in an ant brain." BMC biology. 19,1 254 (2021). doi:10.1186/s12915-021-01188-w
- Zhao, Liangzhen et al. "Analysis of Transcriptome and Epitranscriptome in Plants Using PacBio Iso-Seq and Nanopore-Based Direct RNA Sequencing." Frontiers in genetics. 10 253 (2019). doi:10.3389/fgene.2019.00253

 Sample Submission Guidelines
Sample Submission Guidelines


