Iso-Seq technology, developed by Pacific Biosciences, is a full-length transcriptome sequencing method grounded in PacBio single molecule real-time sequencing (SMRT) technology. It directly sequences full-length cDNA without fragmenting RNA molecules, enabling the generation of high-quality full-length transcript sequences that can extend up to 10 kb or even longer. The key advantage of Iso-Seq lies in its ability to offer complete transcript information, encompassing the 5' untranslated region (5'UTR), 3' untranslated region (3'UTR), and polyA tail. This circumvents the complex processes of transcript splicing and reconstruction inherent in traditional RNA-seq technology.
Iso-Seq can precisely identify biological phenomena such as alternative splicing events, alternative start and stop sites, fusion genes, and long noncoding RNAs (lncRNAs). This provides a novel perspective for investigating gene expression regulation, transcriptome complexity, and gene function. Moreover, due to its capacity to cover full-length transcripts, Iso-Seq is particularly well-suited for studying species without a reference genome. It can uncover transcript isomers and new genes that traditional RNA-seq methods have failed to detect.
Overview Importance of Iso-Seq in Biology
Iso-Seq technology holds significant importance in genomics and transcriptomics research because of its full-length transcript sequencing capabilities. It not only enhances the accuracy of genome annotation but also reveals the complexity of the transcriptome, promotes the discovery of new genes and transcripts, and offers a valuable tool for studying non-reference genome species.
Improving the accuracy of genome annotation: Iso-Seq technology generates full-length transcripts, allowing for the direct determination of exon boundaries, splicing sites, and alternative splicing sites. This leads to more accurate gene annotation. For instance, in plant transcriptome studies, the full-length transcripts obtained through Iso-Seq have significantly enhanced the refinement of gene models. Additionally, Iso-Seq can identify fusion genes and methylation sites, further augmenting the information available for genome annotation.
Revealing the complexity of transcriptome: Iso-Seq technology enables a comprehensive analysis of the intricate transcriptome structure, including alternative splicing events, non-coding RNAs, mRNA degradation (NMD), and polyadenylation. In mammalian studies, for example, Iso-Seq has uncovered numerous new genes and transcripts that were not covered by the reference transcriptome, highlighting its superiority in exploring transcriptome complexity.
Promote the discovery of new genes and new transcripts: Iso-Seq's ability to generate full-length sequences provides a distinct advantage in the discovery of new genes and transcripts. In soybean research, Iso-Seq data covered over 80% of the gene sites identified by RNA-Seq and discovered many novel genes. Moreover, Iso-Seq has been used to detect the expression of pseudogenes in mammals, shedding light on the role of pseudogenes in developmental dynamics.
Applications in Genomics
Gene discovery and annotation
a) Identification of novel genes and isoforms
Gene discovery is a fundamental task in genomics research. Modern bioinformatics software empowers researchers to efficiently identify genes from genome sequences, especially in prokaryotes such as bacteria, archaea, and viruses, where the process is relatively straightforward and accurate.
Artificial intelligence technologies, particularly deep learning algorithms, play a crucial role in genome annotation. These algorithms enhance the accuracy and speed of gene prediction. For example, by integrating statistical information with comparisons to known coding sequences, the dual genome algorithm can precisely locate new gene regions.
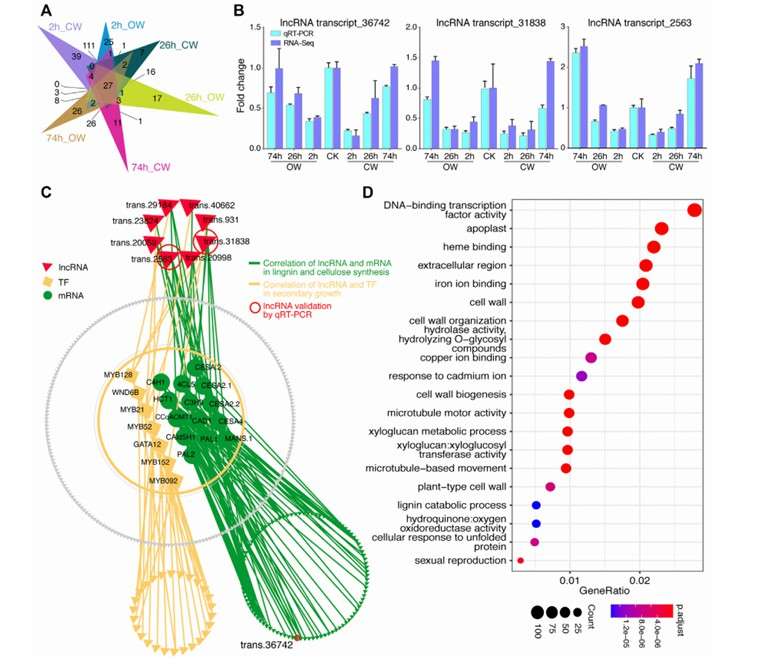 Differential expression analysis of lncRNA (Zhang et al., 2022)
Differential expression analysis of lncRNA (Zhang et al., 2022)
b) Refining gene models
Gene annotation involves not only pinpointing the location of genes but also accurately defining their internal structures, such as introns and exons. This process typically requires the integration of various computational methods, including sequence comparisons with known gene databases and the optimization of annotation results using machine learning algorithms. Genome-wide annotation is especially vital after the completion of genome sequencing for new species, as it provides a foundation for subsequent functional research.
Services you may interested in
Want to know more about the details of Iso-seq? Check out these articles:
Comparative genomics
a) Cross-species analysis of gene structures
Comparative genomics involves comparing genome sequences across different species to uncover evolutionary relationships and conserved elements. This approach not only helps identify similarities and differences between species but also aids in understanding the expansion and contraction of gene families. For example, by analyzing the coding regions of BPI genes in multiple species, researchers can explore their genetic diversity and functional disparities across different organisms.
b) Evolutionary insights
Comparative genomics offers a powerful tool for understanding species evolution. Analyzing the TCTP protein across different species, researchers have elucidated its functional differentiation and evolutionary trajectory. Gene replication mechanisms, such as crossover, inversion, and chromosome multiplication, also play a significant role in evolution, facilitating the emergence of new functions. By comparing genome data from different species, researchers can explore the impact of genome selection on trait variation.
Gene discovery, annotation, and comparative genomics are widely applied in modern life science research. In fields such as medicine, agriculture, and environmental science, researchers can better understand gene expression patterns, disease-related genetic variations, and the genetic basis of complex traits through genome annotation and comparative analysis. With the advancement of high-performance computing and cloud computing technologies, the processing of large-scale genome data has become more efficient, further propelling research in these areas. These aspects of genomics research not only drive the progress of basic science but also provide crucial support for clinical diagnosis, drug development, and biotechnology applications.
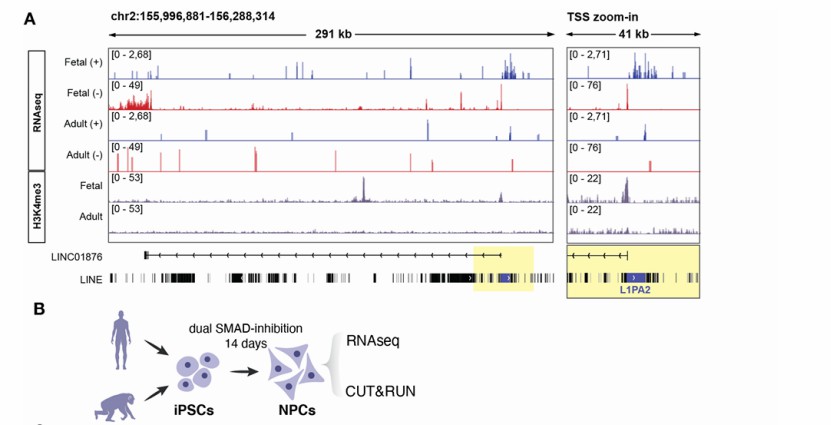 The L1-lncRNA LINC01876 is a human-specific transcript (Raquel et al., 2023)
The L1-lncRNA LINC01876 is a human-specific transcript (Raquel et al., 2023)
Applications in Transcriptomics
A. Alternative splicing studies
a) Identification of splicing variants
Alternative splicing is a process by which a single gene generates multiple mRNA isoforms through different splicing patterns, thereby increasing the diversity of gene expression. High-throughput sequencing technologies, such as RNA-Seq, provide a robust means for comprehensively studying alternative splicing. rMATS, for example, is a statistical method used to detect and quantify differential alternative splicing in repetitive RNA-Seq data. It can handle paired samples, such as case-control studies, and account for individual variation. Tools like IsoformSwitchAnalyzeR can also predict splicing variants by calculating changes in splicing sites.
b) Functional implications of alternative isoforms
Alternative splicing not only adds complexity to the transcriptome but can also influence protein function. Some alternative isoforms may possess specific biological functions, such as regulating gene expression. In cancer research, alternative splicing has been shown to contribute to disease development by altering protein structure and function. Additionally, studies have revealed that alternative splicing events are highly tissue-specific and can significantly impact cell function.
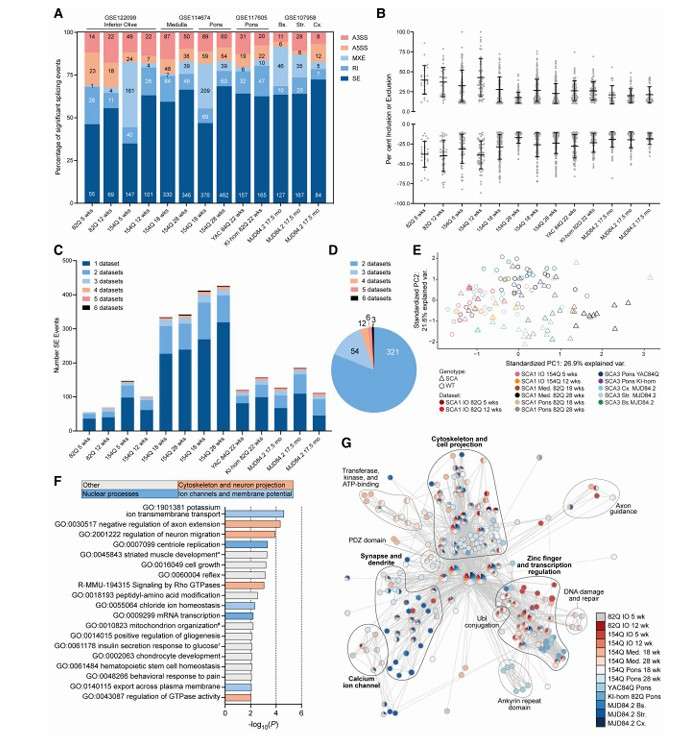 Alternative splicing is dysregulated across affected brain regions in CAG expansion SCA mouse models (Hannah et al., 2024)
Alternative splicing is dysregulated across affected brain regions in CAG expansion SCA mouse models (Hannah et al., 2024)
B. Transcriptome profiling
a) Quantitative analysis of isoform expression
A key application of transcriptome analysis is the quantitative assessment of isoform expression levels. This typically relies on RNA-Seq data and is achieved by identifying reads across splicing sites. The MISO algorithm, for example, can measure the relative expression levels of different isoforms resulting from exon skipping events. A range of computational resources and statistical methods have been developed to calculate transcriptome and isoform diversity.
b) Differential isoform usage across conditions
Alternative splicing exhibits significant differences in isoform usage under different conditions. During development, for instance, the abundance of alternative splicing events increases substantially. Some studies have also found that alternative splicing undergoes significant changes in disease states, such as cancer, which may offer new biomarkers for disease diagnosis and treatment.
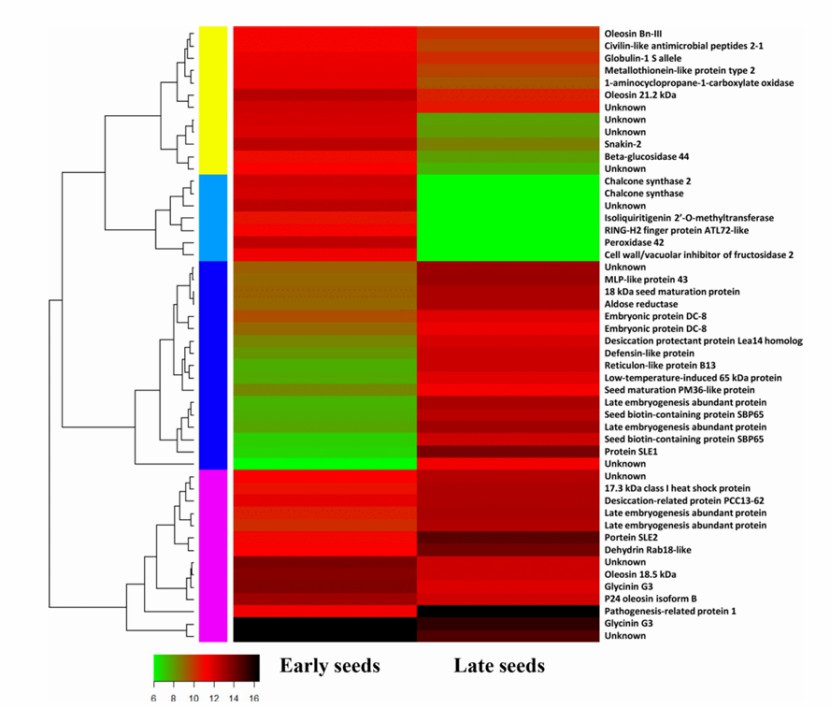 Heat map of top 50 differentially expressed genes between early- and late-stages of seed development in S. tora (Kang et al., 2020)
Heat map of top 50 differentially expressed genes between early- and late-stages of seed development in S. tora (Kang et al., 2020)
Alternative splicing and transcriptome analysis are essential tools in modern biological research. They not only uncover the complexity of gene expression but also provide a new perspective for understanding gene function and disease mechanisms. By combining high-throughput sequencing technology with advanced bioinformatics tools, researchers can conduct more comprehensive analyses of alternative splicing and its changes under various conditions, thereby promoting the development of basic and clinical research.
Case Studies of Iso-Seq
Successful Iso-Seq applications in published studies
Transcriptome research in plants: Iso-Seq technology has demonstrated remarkable advantages in plant transcriptome studies. Through PacBio Iso-Seq, researchers can generate full-length transcripts, avoiding the issues associated with transcriptome reconstruction caused by the short read lengths of traditional RNA-Seq. This enables Iso-Seq to perform well in gene annotation, new gene discovery, long noncoding RNA (lncRNA) identification, and splicing isomer detection.
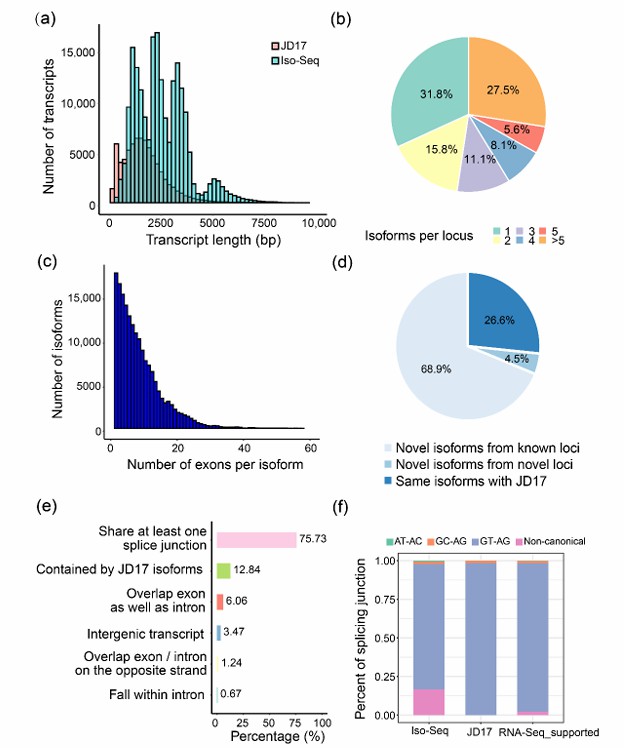 Summary of the Iso-Seq data (Liu et al., 2022)
Summary of the Iso-Seq data (Liu et al., 2022)
Improvement of genome annotation: In the study of model plants like Arabidopsis thaliana, reanalyzing the genome annotation of TAIR10 using Iso-Seq technology successfully recovered nearly 60% of the gene model and identified new gene loci. This result highlights Iso-Seq's ability to significantly enhance the accuracy and completeness of genome annotation.
Transcriptome analysis of non-model plants: Research on Halogeton glomeratus has shown that Iso-Seq technology can provide complete transcriptome data and uncover the complex molecular mechanisms of plant salt stress responses. This is of great significance for the breeding of salt-tolerant crops.
Application in cancer research: Iso-Seq technology is extensively used in cancer research. By sequencing full-length RNA, it can reveal the heterogeneity of tumor cells and gene expression patterns. For example, combining the Kinnex full-length RNA kit with Iso-Seq technology enables researchers to obtain high-quality cancer transcriptome data, offering new insights for cancer treatment.
Agricultural biotechnology: In wheat research, integrating TeloPrime technology with Iso-Seq sequencing has enabled researchers to construct a high-quality wheat hexaploid genome annotation library. This provides crucial support for future wheat gene function research and breeding.
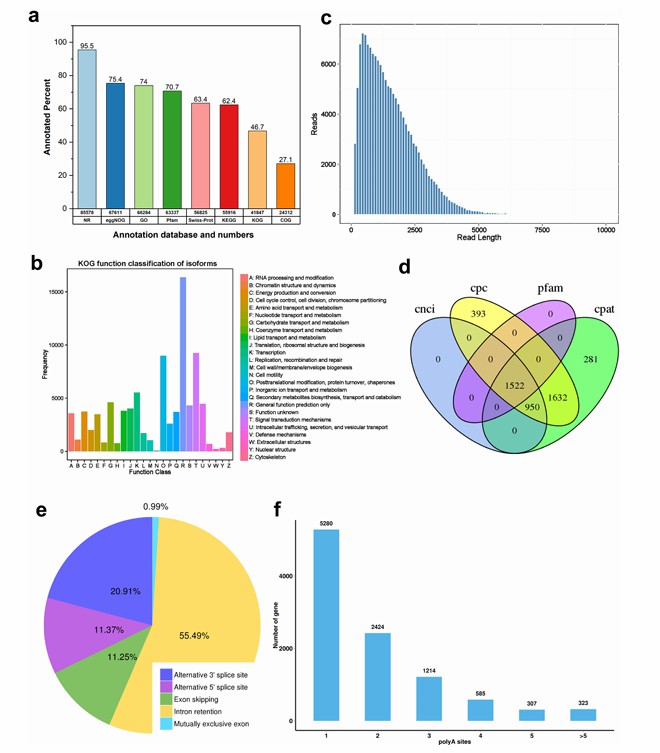 Prediction of functions of Iso-Seq isoform in G. purpurascens (Abdul et al., 2024)
Prediction of functions of Iso-Seq isoform in G. purpurascens (Abdul et al., 2024)
Impact on biomedical research and plant breeding
The applications of Iso-Seq in biomedical research and plant breeding have had a significant impact on these fields. In biomedical research, Iso-Seq has the potential to identify new biomarkers and therapeutic targets for a variety of diseases, including cancer, neurological disorders, and autoimmune diseases. By providing a more complete and accurate picture of the transcriptome, Iso-Seq can help researchers understand the molecular mechanisms underlying disease development and progression.
Biomedical research: In biomedical research, Iso-Seq has the potential to identify new biomarkers and therapeutic targets for various diseases, including cancer, neurological disorders, and autoimmune diseases. By providing a more complete and accurate view of the transcriptome, Iso-Seq helps researchers understand the molecular mechanisms underlying disease development and progression. Sequencing full-length RNA allows for more precise identification of disease-related splicing variants, fusion genes, and mutation sites, which is beneficial for understanding disease mechanisms and providing data support for personalized medicine.
Plant breeding: In plant breeding, Iso-Seq technology helps researchers better understand the genetic regulatory networks of plants by providing comprehensive transcriptome data. In cotton research, analyzing the dynamic changes of the transcriptome under different abiotic stresses has revealed the response mechanisms of cotton to drought, salinity, and other stresses, providing important molecular markers for breeding.
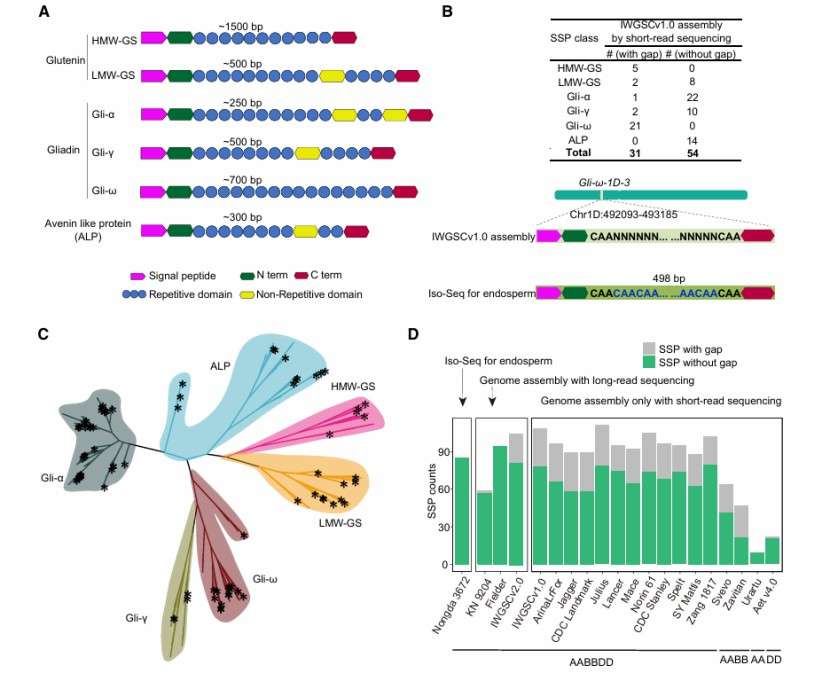 Iso-Seq fills gaps in transcripts encoding seed-storage proteins (Zhang et al., 2024)
Iso-Seq fills gaps in transcripts encoding seed-storage proteins (Zhang et al., 2024)
Improving the accuracy of genome annotation: Iso-Seq technology significantly improves genome annotation accuracy by generating full-length transcripts. In soybean (Glycine max) research, Iso-Seq revealed over 80% of gene loci and detected numerous allelic variations, laying the foundation for soybean genetic improvement and functional gene research.
Promote the development of plant functional genomics: The application of Iso-Seq technology has greatly advanced the development of plant functional genomics. In rice research, combining short-read RNA-Seq and Iso-Seq data has enabled researchers to construct a more comprehensive gene expression map, providing an important reference for rice molecular breeding.
Promote the protection of agricultural biodiversity: Iso-Seq technology also plays a crucial role in the protection of agricultural biodiversity. In mango research, analyzing genome association mapping using Iso-Seq technology helps researchers better understand the genetic basis of fruit quality and stress resistance, providing a scientific basis for mango genetic improvement.
Conclusion
Iso-Seq represents a formidable advancement in the realms of genomics and transcriptomics, offering unparalleled capabilities for the identification of novel genes and isoforms, the refinement of gene models, and the detailed analysis of gene structures across diverse species. Within genomics, Iso-Seq facilitates the discovery of new genetic elements and enhances the accuracy of gene annotations.
In the sphere of transcriptomics, it provides crucial insights into alternative splicing mechanisms and the regulatory landscapes of gene expression. Furthermore, Iso-Seq has increasingly been applied to plant research, oncology, and agricultural biotechnology, where it unveils complex pathways of gene regulation, elucidates tumor characteristics, and assists in the construction of high-fidelity genome annotations.
Looking to the future, the integration of Iso-Seq with additional omics datasets promises to yield comprehensive biological insights. Its potential clinical applications, particularly in the realms of disease diagnosis and therapeutic interventions, appear promising. Additionally, Iso-Seq's utility in promoting sustainable plant breeding marks a significant step forward. As such, Iso-Seq is well-positioned to make substantial contributions to the field of scientific research in the foreseeable future.
References:
- Nam V. Hoang, Agnelo Furtado, Patrick J. Mason, et al. "A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing." BMC Genomics (2017) 18:395. DOI 10.1186/s12864-017-3757-8
- Zhang Zekun, Wang Huiyuan, Wu Ji, et al. "Comprehensive Transcriptome Analysis of Stem-Differentiating Xylem Upon Compression Stress in Cunninghamia Lanceolata." Frontiers in Genetics (2022): 843269. www.frontiersin.org
- An Dong, Hieu X. Cao, Li Changsheng, et al. "Isoform Sequencing and State-of-Art Applications for Unravelling Complexity of Plant Transcriptomes." Genes (2018): 9. http://dx.doi.org/10.3390/genes9010043
- Khaled Moustafa, Joanna M. Cross, et al. "Genetic Approaches to Study Plant Responses to Environmental Stresses: An Overview." Biology (2016) 5:20. doi:10.3390/biology5020020
- Matthias Lienhard, TwanvandenBeucken, BerndTimmermann, et al. "IsoTools: a flexible workflow for long-read transcriptome sequencing analysis." Bioinformatics (2023) 39: 6. https://doi.org/10.1093/bioinformatics/btad364
- Raquel Garza, Diahann Atacho, Anita Adami, et al. "L1 retrotransposons drive human neuronal transcriptome complexity and functional diversification." bioRxiv (2023): 6. https://doi.org/10.1101/2023.03.04.531072
- Sang-Ho Kang, So-Ra Han, Woo-Haeng Lee, et al. "De novo transcriptome sequence of Senna tora provides insights into anthraquinone biosynthesis." PLOS ONE (2020) 5:7. https://doi.org/10.1371/journal.pone.0225564
- Abdul Rehman, Chunyan Tian, Shoupu He, et al. "Transcriptome dynamics of Gossypium purpurascens in response to abiotic stresses by Iso-seq and RNA-seq data." Scientific Data (2024) 11: 477. https://doi.org/10.1038/s41597-024-03334-9
- Liu Jing, Chen Shengcai, Liu Min, et al. "Full-Length Transcriptome Sequencing Reveals Alternative Splicing and lncRNA Regulation during Nodule Development in Glycine max." Int. J. Mol. Sci. (2022) 23: 7371. https://doi.org/10.3390/ijms23137371
- Zhang Zhaoheng, Liu Dan, Li Bingyong, et al. "Ak-mer-based pangenome approach for cataloging seed-storage-protein genes in wheat to facilitate genotype-to-phenotype prediction and improvement of end-use quality." Molecular Plant (2024): 1038-1053. https://doi.org/10.1016/j.molp.2024.05.006
- Hannah K. Shorrock, Claudia D. Lennon, Asmer Aliyeva, et al. "Widespread alternative splicing dysregulation occurs presymptomatically in CAG expansion spinocerebellar ataxias." BRAIN (2024): 486-504. https://doi.org/10.1093/brain/awad329

 Sample Submission Guidelines
Sample Submission Guidelines


