
 Sample Submission Guidelines
Sample Submission Guidelines
Step-by-Step Guide to Iso-Seq Protocol: From Sample Preparation to Data Analysis
Isoform sequencing (Iso-Seq) is a single-molecule real-time sequencing technology developed by PacBio, which is used for the analysis and functional characterization of full-length transcripts. This technology can provide high-quality full-length transcript sequence data by directly sequencing RNA molecules without transcriptome assembly. This method is especially suitable for gene annotation, isomer discovery, alternative splicing, polyadenylation (APA) research and gene fusion detection.
Introduction of Iso-Seq
Iso-Seq is a single molecule real-time sequencing technology based on PacBio platform, which is used for sequencing and analyzing full-length transcripts. Its core steps include the following key links:
Transformation from RNA to cDNA: First, RNA samples are transformed into cDNA, which is the basis of subsequent sequencing.
Construction of cDNA library: Insert cDNA fragments into library vectors suitable for sequencing, such as SMRTbell library format.
Sequencing: Sequel system is used for sequencing to generate high-quality long reading data.
Generating circular consensus sequence (CCS): Processing the original sequencing data by advanced algorithm of PacBio to generate high-quality circular consensus sequence.
Data analysis: Using special software tools (such as SMRT Link or TAGET), analyze the generated data, including identifying transcript isomers, splicing variations, polyadenylation sites and so on.
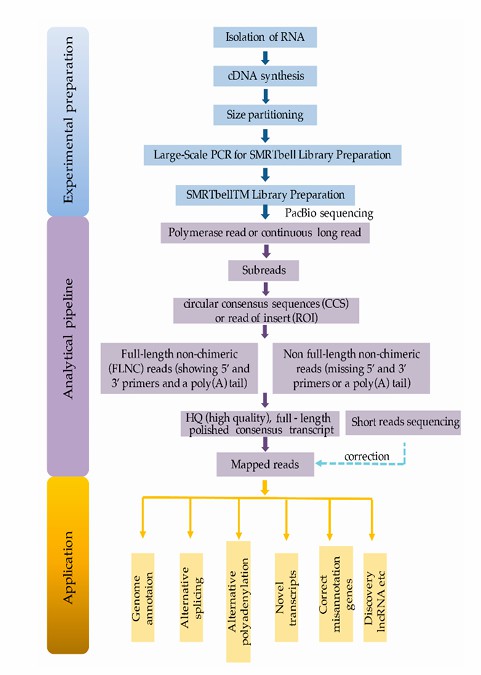 Shematic protocol of Iso-Seq (An et al., 2018)
Shematic protocol of Iso-Seq (An et al., 2018)
Importance of a comprehensive guide for Iso-Seq experiments
Because Iso-Seq experiment involves many steps, from sample preparation to data analysis, each step needs precise operation and optimization. Therefore, it is very important to provide a comprehensive experimental guide to ensure the success of the experiment and the reliability of the results. For example, RNA quality control, optimization of library construction and adjustment of sequencing parameters need to be considered during sample preparation.
In terms of data analysis, PacBio recommended using SMRT Link software for polyploidy and isomer analysis, and further functional characterization by combining with community tools such as SQANTI, TAMA and LoReAn. In addition, for different types of samples (such as single cells or long reading samples), it is necessary to select appropriate kits and analysis procedures.
In order to improve the efficiency of data analysis and reduce errors, researchers need a systematic process to deal with Iso-Seq data. This includes quality control, splicing and annotation of original sequencing data, differential expression analysis and functional annotation. In addition, with the deepening of research, how to integrate Iso-Seq data with other omics data (such as RNA-Seq and protein omics data) to obtain more comprehensive biological insights is also an important direction of current research.
Services you may interested in
Want to know more about the details of Iso-seq? Check out these articles:
Step-by-Step Iso-Seq Protocol
Iso-seq process is mainly to extract high-quality RNA from samples, then reverse transcribe to synthesize full-length cDNA, then fragment the cDNA, then connect it to sequencing adapter to construct sequencing library, then load the library into PacBio sequencer, obtain long reading sequence containing complete transcript information by using single molecule real-time sequencing technology, and finally use special analysis software for sequence correction, clustering and annotation to accurately identify and identify different transcript isomers.
A. Sample preparation
- RNA extraction and quality control
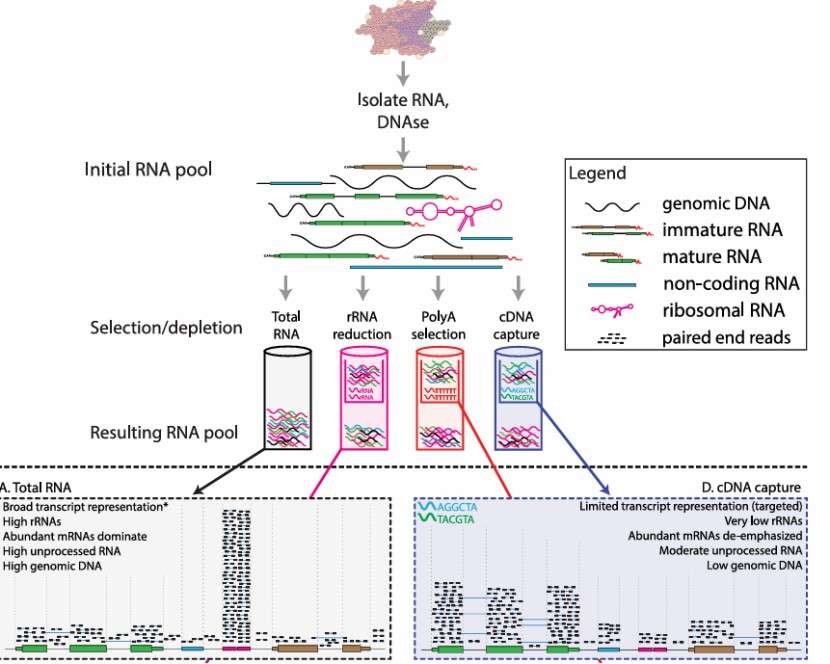
a) Methods and tools: RNA extraction usually adopts phenol chloroform method, silica gel column method or magnetic bead method. Phenol chloroform method is simple but easy to be polluted, silica gel column method has high purity but high cost, and magnetic bead method combines the advantages of rapidity, automation and high recovery. In addition, commonly used methods include the use of commercial kits, such as the Qiagen RNeasy kits, which are based on silica-membrane technology. These kits can efficiently isolate total RNA from various sample types, including tissues, cells, and blood. For plant samples, the CTAB (Cetyltrimethylammonium bromide) method is often employed, which is effective in dealing with the high levels of polysaccharides and polyphenols present in plant tissues. Tools like centrifuges are essential for separating the phases during extraction, and spectrophotometers are used to measure the concentration of the extracted RNA.
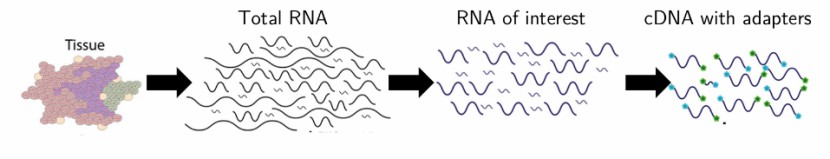 The workflow of RNA extraction (Griffith et al., 2015)
The workflow of RNA extraction (Griffith et al., 2015)
b) Quality evaluation: RNA quality can be evaluated by measuring A260/A280 ratio (ideal range is 2.0-0.2) and A260/A230 ratio (ideal range is 2.0-0.2) with spectrophotometer. The quality of the extracted RNA is vital for downstream applications. The RNA Integrity Number (RIN) is a widely used metric to assess RNA quality. It ranges from 1 to 10, with 10 indicating the highest quality. RIN values are determined using automated capillary electrophoresis systems, such as the Agilent 2100 Bioanalyzer. An RNA sample with a RIN value of 7 or higher is generally considered suitable for Iso-Seq. Samples with lower RIN values may result in degraded RNA, leading to incomplete or inaccurate sequencing results.
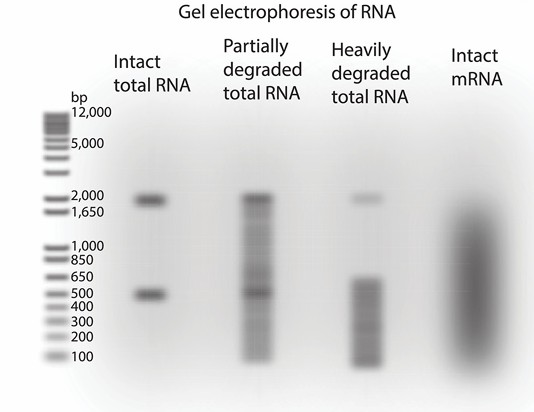 Quality control and size selection of RNA (Griffith et al., 2015)
Quality control and size selection of RNA (Griffith et al., 2015)
- cDNA synthesis
a) Reverse transcription method: Using Smarter Total RNA-SeqKit (Clontech) or Kapa Hifi RNA-to-cDNA Synthesis Kit to synthesize cDNA. These methods reverse transcribe RNA into cDNA by random primers or oligo (dT) primers, and improve the amplification efficiency by using KAPA HiFi enzyme. Random hexamer priming is a more general approach that can bind to any region of the RNA molecule, making it suitable for a wide range of transcripts. Gene-specific priming is used when only specific transcripts are of interest. Reverse transcriptase enzymes, such as Superscript III, are commonly used in these reactions.
 Schematic synthesis of cDNA (Griffith et al., 2015)
Schematic synthesis of cDNA (Griffith et al., 2015)
b) cDNA quality inspection: After cDNA synthesis, it is important to perform quality checks. This can be done by running the cDNA on an agarose gel to check for the presence of a smear or specific bands, indicating successful synthesis. Additionally, quantitative PCR (qPCR) can be used to assess the quantity and integrity of the cDNA. The cDNA should have a consistent concentration and be free from inhibitors that could affect downstream library preparation and sequencing steps. Check the size distribution of cDNA fragments to ensure that they are suitable for subsequent library construction.
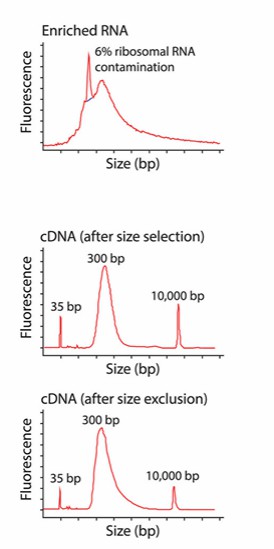 Different size distribution of cDNA before and after size selection (Griffith et al., 2015)
Different size distribution of cDNA before and after size selection (Griffith et al., 2015)
B. Library construction
- PacBio library construction
a) Size selection and amplification: For PacBio library construction, size selection is an important step. This can be achieved using techniques such as gel extraction or the use of size selection beads. The goal is to isolate cDNA fragments within a specific size range, typically around 1-10 kb for full-length transcript analysis. Then the library was amplified by KAPA HiFi PCR to increase the library concentration.The primers used in this amplification step are designed to add the necessary adapter sequences for sequencing on the PacBio platform.
b) Quality control step: Quality control of the constructed library is essential. This includes measuring the concentration of the library using methods like Qubit fluorometry, which provides a more accurate measurement of the DNA concentration compared to spectrophotometry. Additionally, the library is analyzed using capillary electrophoresis or other high-throughput methods to check for the presence of the correct size distribution of fragments. Any libraries that do not meet the quality standards may need to be reamplified or reconstructed.
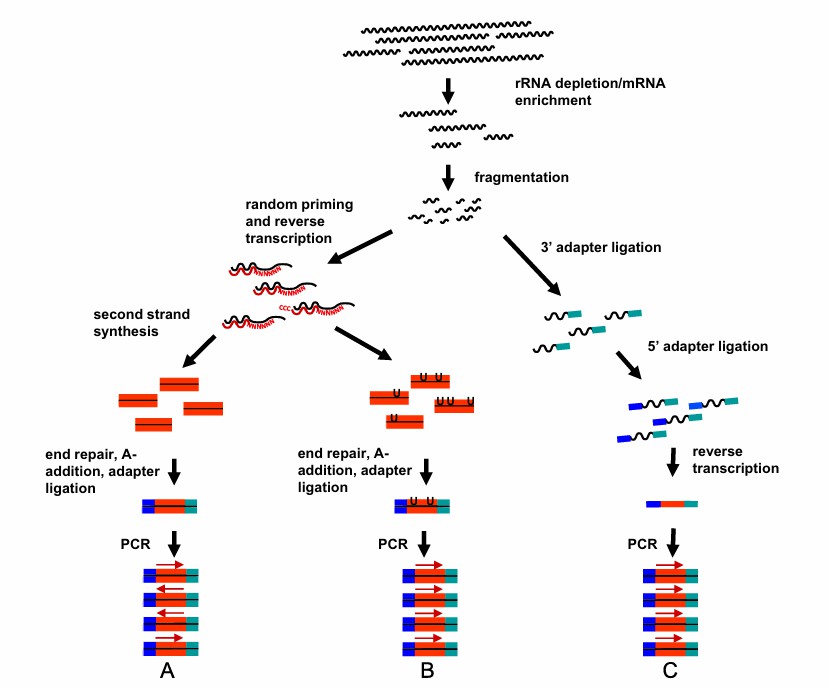 The most common Iso-Seq library construction methods (Erwin et al., 2014)
The most common Iso-Seq library construction methods (Erwin et al., 2014)
- Sequencing run settings
a) SMRT cell loading: SMRT cell is the platform for sequencing on the PacBio system. Loading the SMRT cell involves carefully adding the prepared library to the cell, ensuring that the correct concentration and volume are used. The library is mixed with sequencing reagents, including polymerase and nucleotides, before being loaded into the cell, and each cell is loaded with about 100-200 ng of library.
b) Sequencing parameters: Several sequencing parameters need to be set up, such as the movie length, which determines how long the sequencing run will last. Longer movie lengths can increase the coverage and the likelihood of obtaining full-length transcripts, but they also increase the cost and time of the experiment. Other parameters include the polymerase binding time and the temperature settings, which are optimized to ensure accurate and efficient sequencing. PacBio Sequel II platform is recommended for sequencing, and the sequencing time is set at 180 minutes, so as to obtain high-quality long reading data.
C. Data generation and quality control
- Original data output and preliminary inspection
a) After the sequencing run, raw data is generated in the form of signal files. These files need to be processed to obtain the sequence reads. Initial checks of the raw data include assessing the signal quality, the number of reads generated, and the distribution of read lengths. Any obvious signs of poor signal quality or low read counts may indicate issues with the sequencing run or the library preparation.
- Error correction and preprocessing
a) Error correction is a necessary step in Iso-Seq data processing due to the relatively high error rate of the PacBio sequencing technology. Tools like Canu and Falcon are commonly used for error correction. These tools use algorithms to correct the sequencing errors by comparing multiple reads of the same region. After error correction, preprocessing steps such as adapter trimming and filtering of low-quality reads are carried out. Software like BBDuk can be used for adapter trimming, and tools like FastQC can be used to assess the quality of the preprocessed reads.
D. Data analysis
- Transcriptome assembly and isomer recognition
a) Recommended process and tools: Transcript assembly is the process of putting together the corrected reads to form complete transcripts. Pipelines like the PacBio Iso-Seq pipeline are designed specifically for this purpose. Tools such as StringTie and Cufflinks can also be used for transcript assembly. After assembly, isoform identification is carried out to identify different splice variants of the same gene. These tools use algorithms to compare the assembled transcripts and identify regions of alternative splicing.
- Functional annotation and alternative splicing analysis
a) Bioinformatics resources and databases: Functional annotation of the identified transcripts involves assigning biological functions to them. This can be done using resources such as the Gene Ontology (GO) database, which provides information on the molecular function, biological process, and cellular component of genes. The Kyoto Encyclopedia of Genes and Genomes (KEGG) database is also useful for understanding the metabolic pathways and biological processes in which the genes are involved. For alternative splicing analysis, databases like the Human Splicing Database (HSD) can be used to compare the identified splicing events with known splicing patterns.
E. Data interpretation and visualization
- A tool for visualizing Iso-Seq results
a) Several tools are available for visualizing Iso-Seq results. Integrative Genomics Viewer (IGV) is a popular tool that can display the aligned reads, transcripts, and gene models. It allows researchers to visualize the alternative splicing events, the coverage of the reads, and the distribution of transcripts across different genes. Another tool, Circos, can be used to create circular visualizations of the data, which are useful for comparing multiple samples or for visualizing the relationships between different genes.
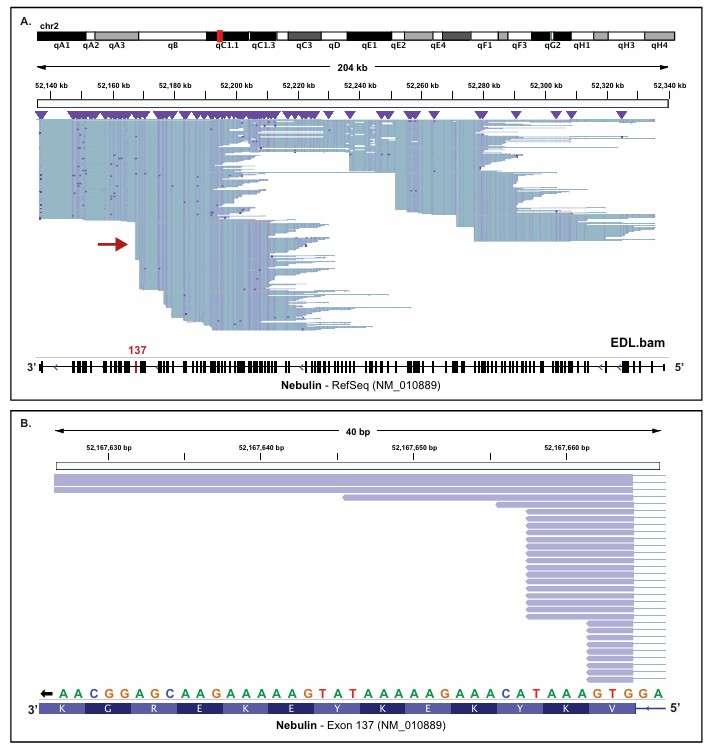 IGV screenshots of reads due to internal oligo-dT priming (Prech et al., 2020)
IGV screenshots of reads due to internal oligo-dT priming (Prech et al., 2020)
- Reporting and sharing Iso-Seq data
a) Once the data analysis is complete, it is important to report and share the Iso-Seq data. This can be done by publishing the results in scientific journals, along with the raw data deposited in public databases such as the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA). Additionally, researchers can share their analysis scripts and processed data on platforms like GitHub, which allows other researchers to reproduce the analysis and build on the results. Clear and detailed reporting of the methods, results, and data sources is essential for the reproducibility and transparency of the research.
The above steps cover the complete Iso-Seq experimental process from sample preparation to data analysis. Each step combines the current mainstream technologies and tools to ensure the efficiency and accuracy of the experiment. For example, RNA extraction uses commercial kits to ensure purity and integrity. cDNA synthesis uses efficient SMARTer kit. Library construction combines size selection and amplification techniques. In the data analysis part, it is recommended to use tools such as LoRC and GMAP to assemble and annotate high quality transcripts. These steps provide a solid foundation for in-depth analysis of gene expression and transcriptome complexity.
Troubleshooting and Tips for Iso-Seq
Although Iso-Seq technology has obvious advantages, there are still many problems. During sample preparation, the quality of RNA extraction is unstable, which is easily affected by factors such as pollution, degradation or low purity, thus reducing the quality of sequencing data. In sequencing, it is common that the sequencing depth is insufficient, and it is difficult to obtain enough full-length transcriptome information because of short reading length or small sample size. In data analysis, it is difficult to detect isomers, which is limited by the incompleteness of genome reference sequence and the low expression level of isomers.
Common problems in Iso-Seq experiment
- Low data quality: The quality of RNA extracted from samples is not high, the degradation is serious or the purity is low, resulting in fewer high-quality fragments in sequencing data. Optimize RNA extraction process to ensure high integrity and purity of RNA. For example, RNA was extracted using a high-purity TRIzol kit, and RNA integrity was checked by agarose gel electrophoresis.
- Insufficient sequencing depth: The short reading length of sequencing or insufficient sample size made it impossible to obtain enough full-length transcriptome information. Increase the sample size or extend the sequencing time to improve the sequencing depth. For example, the Illumina platform is used for at least 10G data sequencing.
- Isomer detection is difficult: Because the genome reference sequence is incomplete or the expression level of isomers is low, it is difficult to identify isomers accurately. Combined with full-length sequencing data and RNA-seq data, more complex algorithms (such as Markov chain-based model) are used to predict and analyze isomers.
- High data processing complexity: The analysis of full-length transcriptome data requires high-performance computing resources and complex bioinformatics tools. Use special software packages (such as SMRTLink of PacBio or TruSight of Illumina) for data processing, and use cloud computing platform (such as Hadoop) to improve computing efficiency.
- High database dependence: High-quality genome annotation database is the basis of full-length transcriptome analysis, but the existing databases are often not comprehensive enough. Build or update reference genome database, such as PISO database, to support full-length transcriptome analysis of more species.
Best practices for successfully running Iso-Seq experiments
- Use high-quality RNA samples to avoid degradation or pollution. It is suggested to use TRIzol kit to extract RNA and verify the integrity of RNA by agarose gel electrophoresis. Fully homogenize the sample to ensure complete cell lysis.
- Select the appropriate sequencing platform according to the research needs. For example, full-length transcriptome sequencing is performed on PacBio platform, or Qualcomm data supplement is performed in combination with Illumina platform. Set a reasonable sequencing depth to ensure that the coverage of each gene is high enough, thus improving the accuracy of full-length transcriptome information.
- Use special software tools for data processing, such as SMRTLink of PacBio, TruSight of Illumina or RNA-seq analysis system based on Hadoop. Combined with many bioinformatics methods, such as DESeq2, NORMSEQ, etc., differential expression analysis and gene annotation were carried out.
- Construct or update reference genome databases, such as PISO database, to support full-length transcriptome annotation and functional analysis. Using the annotation information in the database, the function of newly discovered genes or transcripts is predicted and classified.
- In the experimental design stage, the sample type, processing conditions and biological repetition times are fully considered. For example, multiple biological replications are set under different physiological conditions to improve the reliability of data analysis. Select appropriate experimental models and sample types according to the research purpose, such as model plants, animal models or human samples
Through the above troubleshooting and best practice guidance, the success rate of Iso-Seq experiment can be effectively improved, and high-quality data support can be provided for subsequent research.
Conclusion
Iso-Seq protocol includes five main steps: the transformation from RNA to cDNA, the construction of cDNA to SMRTbell library, sequencing with Sequel system, generating consensus cyclic sequence (CCS) and finding isomers through Iso-Seq analysis. In addition, the technology also supports multiplex sequencing and single cell sequencing, which makes it have unique advantages in the study of complex samples and rare cell types.
Sample preparation is crucial as its quality impacts subsequent sequencing accuracy. High-quality RNA is essential; degradation or pollution can cause sequencing failure. In preparation, adding multiple A-tails and using an rRNA depletion kit following standard procedures is necessary.
Experimental design should be rational, considering sample type, sequencing depth, and target analysis. For plant research, choose tissues and time points based on gene expression. Include multiple strategies for better data coverage and accuracy.
Sequencing parameters like on-board concentration and time need optimization according to sample features and analysis requirements. Use quality instruments and reagents following the manufacturer's guide to enhance data quality.
Data analysis demands specialized tools like SMRT Link or TAGET to handle complex isomer data. Correct potential deviations and verify results with other technologies like RNA-seq. During the experiment, continuously monitor sample and sequencing quality. Check if the data meets expectations post-sequencing and adjust the scheme promptly. Ensure all data is complete after the experiment.
References:
- Othman Al-Dossary, Agnelo Furtado, Ardashir KharabianMasouleh, Bader Alsubaie, Ibrahim Al‑Mssallem and Robert J. Henry. "Long read sequencing to reveal the full complexity of a plant transcriptome by targeting both standard and long workflows." Plant Methods (2023) 19:112. https://doi.org/10.1186/s13007
- Wang Bo, Elizabeth Tseng, Primo Baybayan, Kevin Eng, Michael Regulski, Jiao Yinping, Wang Liya, Andrew Olson, Kapeel Chougule, Peter Van Buren1 and Doreen Ware. "Variant phasing and haplotypic expression from long-read sequencing in maize." Communications Biology (2020) 3:78. https://doi.org/10.1038/s42003-020-0805-8
- Prech Uapinyoying, Jeremy Goecks, Susan M. Knoblach, Karuna Panchapakesan, Carsten G Bonnemann, Terence A. Partridge, Jyoti K Jaiswal, and Eric P. Hoffman. "A new long-read RNA-seq analysis approach identifies and quantifies novel transcripts of very large genes." bioRxiv (2020) 1:9. https://doi.org/10.1101/2020.01.08.898627
- Sara Ballouz, Alexander Dobin, Thomas R. Gingeras and Jesse Gillis. "The fractured landscape of RNA-seq alignment: the default in our STARs." Nucleic Acids Research (2018): 5125-5138. doi: 10.1093/nar/gky325
- Alexander Dobin1, Carrie A. Davis, Felix Schlesinger, JorgDrenkow, Chris Zaleski, Sonali Jha, PhilippeBatut, MarkChaisson and Thomas R. Gingeras. "STAR: ultrafast universal RNA-seq aligner." Bioinformatics (2013): 15-21. doi:10.1093/bioinformatics/bts635
- Malachi Griffith, Jason R. Walker, Nicholas C. Spies, Benjamin J. Ainscough, Obi L. Griffith. "Informatics for RNA Sequencing: A Web Resource for Analysis on the Cloud." PLOS Computational Biology (2015): e1004393. doi:10.1371/journal.pcbi.1004393
- Erwin L. van Dijka,n, Yan Jaszczyszynb, Claude Thermes. "Library preparation methods for next-generation sequencing: Tone down the bias." Experimental Cell Research (2014): 12-20. http://dx.doi.org/10.1016/j.yexcr.2014.01.008


