
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

Transcriptomics is a crucial field in biological research, dedicated to exploring gene expression and its regulatory mechanisms. At its core lies the comprehensive analysis of gene expression patterns across diverse tissues, cells, or experimental conditions. Traditional short-read and long-RNA sequencing technologies, such as those offered by Illumina, face limitations due to fragmentation. This fragmentation prevents the complete coverage of full-length transcripts, leading to incomplete sequence splicing and inaccurate gene annotation. In contrast, full-length transcript sequencing provides comprehensive information about transcripts, including the 5' and 3' exons, introns, and poly-A tails. This detailed information enables a more in-depth understanding of gene expression and regulatory mechanisms.
Iso-Seq technology is a full-length transcriptome sequencing method based on single molecule real-time (SMRT) sequencing, which was developed by PacBio company. By synthesizing the full-length cDNA library, this technology can directly capture the complete transcript sequence by using its super-long reading ability (the average length can reach 10-15 kb) without relying on the reference genome. Iso-Seq technology has significant advantages in sequencing full-length transcripts, such as:
High Precision in detection: Iso-Seq showcases remarkable accuracy in identifying splicing events, transcript isoforms, fusion genes, and non - coding RNAs. This precision provides researchers with a more nuanced and accurate view of the transcriptome, enabling a deeper understanding of gene function and regulation.
Comprehensive Transcript Coverage: Covering the complete transcript from 5' end to 3' end, including intron and poly-A tail, which is helpful to analyze the complex transcriptome structure.
Versatility in application: It is suitable for full-length transcriptome sequencing of species without reference genome, and supports parallel analysis of multi-tissue samples.
Enhanced effects through interfration: Combining with other sequencing technologies (such as RNA-seq) can further improve the parsing ability of transcriptome data.
PacBio is the main promoter and leader of Iso-Seq technology. Sequel II series, its third-generation sequencing platform, has significantly improved the accuracy and consistency of full-length transcripts through optimized HiFi sequencing technology. PacBio's Iso-Seq technology is not only widely used in academic research, but also used for genome annotation and transcriptome analysis of plants, animals and humans.
In addition, PacBio has developed a variety of supporting tools and software (such as SMRT Link, IsoCon, TAMA, etc.) to provide us with a complete solution from sample preparation to data analysis.
Services you may interested in
Want to know more about the details of Iso-seq? Check out these articles:
Isoform sequencing (Iso-Seq) is a full-length transcript sequencing method based on SMRT technology, which was developed by PacBio. This technology aims to capture and analyze the complete sequences of different isoforms in the genome, thus providing more comprehensive transcriptome information. The following discribes a schematics principle of Iso-Seq.
SMRT: SMRT is a third-generation sequencing technology, which performs sequencing by capturing DNA templates in a Zero-Mode Waveguide, ZMW). This method does not need to interrupt DNA fragments, but directly sequences a single DNA molecule, so as to grow a long reading sequence (usually 10kb or longer), and can completely capture the full-length transcript from the 5' end to the poly-A end.
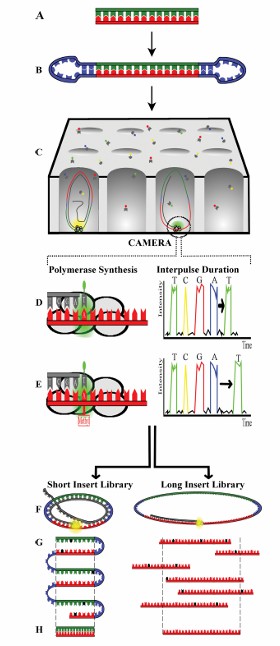 Overview of SMRT sequencing technology (Simon et al., 2018)
Overview of SMRT sequencing technology (Simon et al., 2018)
Capture full-length transcript: Iso-Seq technology can generate high-quality full-length transcript sequences by sequencing cDNA directly without interruption or splicing. These sequences include complete information from the 5' end to the poly-A end, which enables researchers to accurately analyze splicing variants, initiation sites, termination sites and post-transcriptional regulatory events such as alternative splicing and polyadenylation (APA).
PacBio Iso-Seq technology is a full-length transcriptome sequencing method based on SMRT. Its core advantage is that it can sequence full-length transcripts without splicing or inference, thus providing more accurate gene expression and transcript isomer analysis. PacBio's SMRT technology is a third-generation sequencing technology, which generates long reading sequences by monitoring the DNA synthesis process in real time. Its core component is SMRT cell, which is a small consumable containing millions of zero-mode waveguides (ZMWs) for capturing DNA molecules and recording the process of nucleotide addition in real time. This technique can generate sequences as long as 10 kb or even longer, and the accuracy is as high as 99%.
PacBio sequencing is characterized by its high accuracy and long reading capacity. Its average reading length is usually between 8-15 kb, and the longest can reach 40-70 kb. This long reading length makes PacBio especially suitable for studying splicing variants and fusion genes in complex genome regions, repetitive sequences and transcriptome.
Iso-Seq is a bioinformatics tool based on full-length RNA sequencing, which is widely used in many fields. The following are specific applications of Iso-Seq in different fields.
Gene discovery and annotation
Enhancing whole genome annotation: Iso-Seq can generate a complete transcript sequence, which is especially important for new species or incompletely sequenced genomes. By comparing with the reference genome, Iso-Seq can accurately locate exon boundaries, splicing sites and alternative splicing connection sites, thus improving the accuracy of gene annotation.
Uncovering new genes and isoforms: Iso-Seq can detect many uncommented transcripts, including new genes, isomers (such AS, APA) and fusion genes. This provides abundant data support for genome research.
Elevating annotation quality: Compared with traditional expression sequence tag (EST), RNA-Seq and homologous inference methods, Iso-Seq can annotate genes more accurately because of its continuous sequence data.
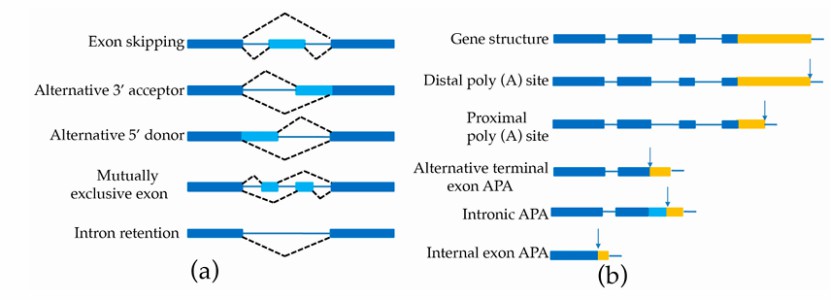 Schematic representation of AS and APA (An et al., 2018)
Schematic representation of AS and APA (An et al., 2018)
Alternative splicing research
Detecting alternative splicing (AS) events: Iso-Seq can directly detect alternative splicing events, including intron retention, exon skipping and exon deletion. These data are helpful to understand the regulation mechanism of gene expression.
Investigating alternative polyadenylation (APA): Iso-Seq can detect APA events in different tissues or conditions, and reveal the post-transcriptional regulation mechanism in different cell types or physiological states.
Quantifying differential splicing: By comparing the transcriptome data of different samples, Iso-Seq can quantitatively analyze differential splicing events and their functional effects, and provide support for disease diagnosis and biomarker development.
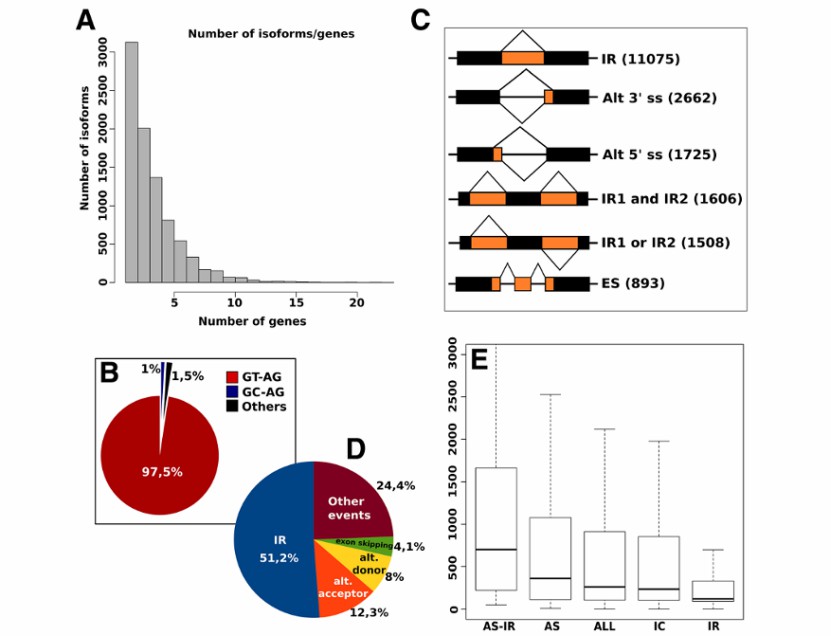 A result sample of alternative splicing research (Nicola et al., 2014)
A result sample of alternative splicing research (Nicola et al., 2014)
Transcriptome and isomer analysis
Simplifying transcriptome assembly and annotation: The full-length sequence data generated by Iso-Seq avoids the complexity of transcriptome assembly and improves the integrity of the transcript, thus more accurately reflecting the gene expression level and post-transcriptional regulation mechanism.
Profiling isoform expression: Using Iso-Seq data, we can accurately analyze the expression patterns of different isomers, including tissue-specific expression and stress response.
Advancing the study on long non-coding RNA (lncRNA): Iso-Seq can detect and annotate lncRNA, which provides an important tool for understanding its role in gene regulation.
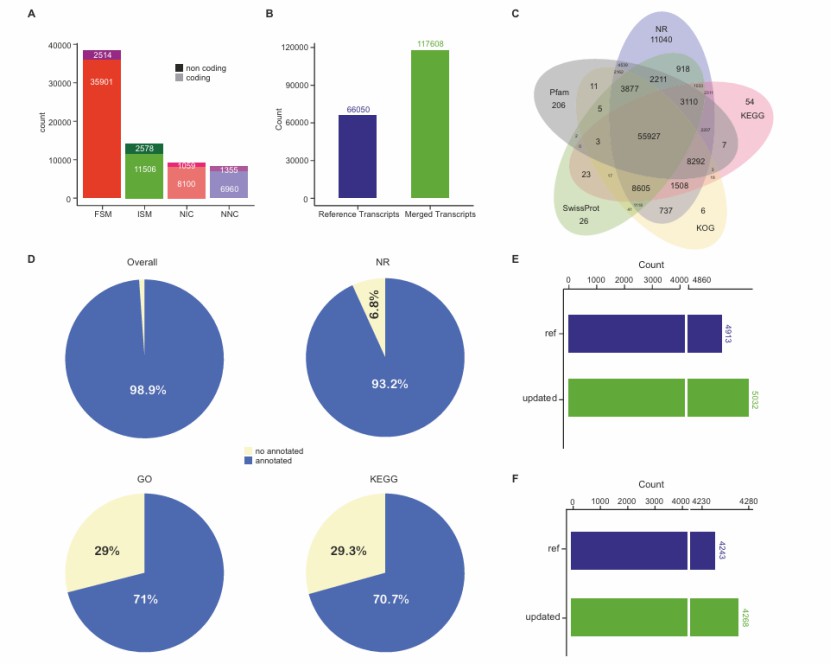 Updating of lettuce reference annotation by HIT-ISOseq (Shi et al., 2024)
Updating of lettuce reference annotation by HIT-ISOseq (Shi et al., 2024)
Comparative genomics
Uncovering inter-species differences: By comparing the Iso-Seq data of different species, species-specific gene expression patterns and post-transcriptional regulation mechanisms can be revealed. For example, studies have shown that Iso-Seq can find unique transcripts and splicing isomers when comparing different plant species.
Tracing evolutionary changes: Iso-Seq data is helpful to analyze the changes of genome structure and function during species evolution, especially in the absence of reference genome.
As an advanced RNA sequencing technology, Iso-Seq has a wide application prospect. It can not only improve the accuracy and completeness of gene annotation, but also deeply study the functions of alternative splicing, transcriptome isomers and long non-coding RNA. In addition, Iso-Seq also shows great potential in comparative genomics and evolutionary research. However, due to the high cost, its large-scale application still faces certain challenges.
For more detailed information, please refer to the articles:
Iso-seq, namely full-length transcript sequencing, is a sequencing method based on PacBio single molecule real-time sequencing technology, and its flow mainly includes the following key links:
Sample preparation
RNA extraction and quality assurance: RNA extraction is a key step in transcriptome sequencing. Usually, specific methods are used to separate RNA molecules from samples and remove unnecessary impurities. For example, DNase I digestion is used to remove DNA contamination, and then RNA is further purified by magnetic beads. The extracted RNA needs quality control, including using Qubit or Agilent Bioanalyzer to detect RNA concentration and integrity.
cDNA synthesis: Reverse transcription of RNA into cDNA (complementary DNA) for subsequent analysis. Commonly used methods include the use of SMARTer Total RNA-Seq cDNA synthesis kit, which can reverse transcribe RNA at the end of poly(A) into cDNA. In the process of cDNA synthesis, special adapter is usually added for subsequent PCR amplification and library construction.
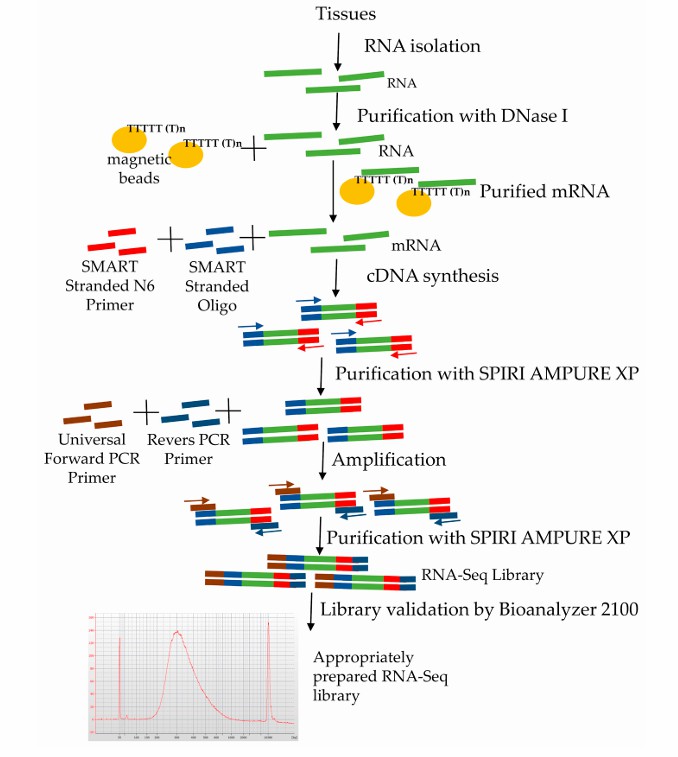 Stages of Iso-Seq library preparation (Marta et al., 2020)
Stages of Iso-Seq library preparation (Marta et al., 2020)
Sequencing and data generation
PacBio library preparation: The library is prepared by connecting cDNA fragments to vectors suitable for sequencing. For example, a library was prepared using the SMARTer P5/P7 linker template and amplified by the KAPA HiFi HotStart PCR system. The amplified library needs quality screening to ensure that it is suitable for sequencing. For example, the library concentration was determined by using Qubit BR DNA HS Assay, and the size was selected by E-gel.
Sequencing operation and data collection: PacBio Sequel II sequencing platform was used for sequencing. During sequencing, multiple SMRT cells can be used for parallel sequencing to improve data coverage. After sequencing is completed, the generated Subreads need to be quality controlled and trimmed to remove low-quality or erroneous sequence fragments.
Data analysis process
Data preprocessing: Quality control was carried out on the original sequencing data to remove low-quality reading segments and linker sequences. Use tools such as Trimmomatic or Cutadapt to trim data to ensure data quality.
Identifying and annotating of full-length transcripts: Using Iso-Seq technology of PacBio, the data are spliced and annotated by SMRT Analysis software (such as SMRT analysis) to generate full-length transcripts (including 5' and 3' UTR regions). The structure and function of transcripts were further analyzed by comparing reference genomes or using de novo assembly method.
Downstream analysis Data: analysis includes differential expression analysis, alternative splicing event detection and transcription initiation site analysis. For example, use tools such as DESeq2 and EdgeR to perform differential expression analysis. Visualization results can be displayed by volcano maps, thermal maps and other methods to help researchers understand the differences of gene expression under different conditions.
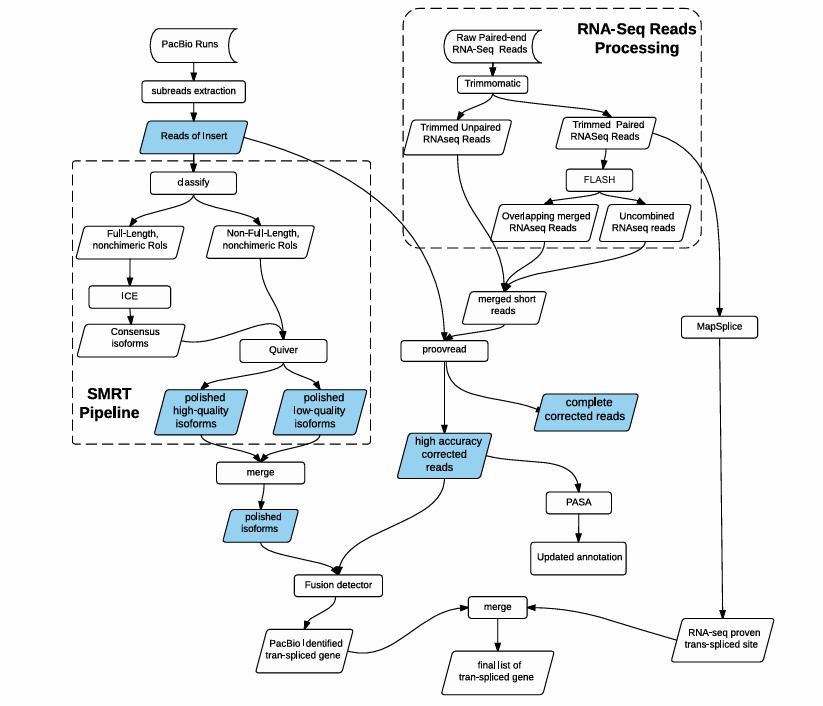 Data processing and analysis pipelines for both RNA-seq data and Iso-Seq data (Jiang et al., 2017)
Data processing and analysis pipelines for both RNA-seq data and Iso-Seq data (Jiang et al., 2017)
Based on the above steps, the complete Iso-Seq protocol includes sample preparation, library preparation, sequencing operation and data analysis. This process can generate high-quality full-length transcript data and provide comprehensive information support for transcriptome research.
For more detailed information, please refer to the article:
The Iso-Seq is a full-length transcript sequencing method based on the third generation sequencing technology, which was developed by PacBio. By providing a complete cDNA sequence, it avoids the step of transcriptome reconstruction in traditional RNA sequencing (such as RNA-seq), thus improving the accuracy and completeness of transcript annotation. Iso-Seq can generate a full-length non-repetitive reading segment (FLNC) of up to 10 kb, covering all regions from the 5' cap to the polyadenylation tail, which makes it a significant advantage in revealing the complexity of transcriptome.
Nevertheless, Iso-Seq also has some limitations, such as higher cost and lower output. However, with the progress of technology and the reduction of cost, Iso-Seq is expected to become an important tool for transcriptome research.
In the future, the development of Iso-Seq technology will have a far-reaching impact on genomics research. The following are several possible development directions and their potential impacts:
Integrating multiple sequencing technologies: Combining short reading and long sequencing technologies (such as RNA-seq and Iso-seq) will help to overcome their respective technical limitations. For example, by integrating short-reading RNA-seq and long-reading Iso-seq data, researchers can more comprehensively analyze the complexity of transcriptome and improve the accuracy of gene annotation.
Application to more species: With the popularization of Iso-Seq technology, its application in non-model species will be more extensive. For example, in the research of marine organisms, insects and plants, Iso-Seq technology will help to reveal the unique genomic characteristics and evolutionary mechanism of these species.
Support personalized medicine: With the deepening of genomics research, Iso-Seq technology may play an important role in personalized medicine. For example, by analyzing the transcriptome data of an individual, a more personalized scheme can be provided for accurate diagnosis and treatment.
Iso-Seq technology is becoming more and more important in transcriptome research, and its future development will greatly promote the progress of genomics research and bring new opportunities to biology, medicine and other fields.
References:


