Amplicon sequencing, a method of targeted DNA sequencing, is broadly utilized for the analysis of genetic variances within defined genomic areas. This technique has become a crucial asset in the field of modern genomics, delivering high levels of accuracy, economic efficiency, and productivity. It is employed across diverse scientific domains, such as in the assessment of microbial biodiversity, the advancement of disease-related research, and the study of evolutionary biological processes.
What is Amplicon Sequencing?
Amplicon sequencing, a form of focused next-generation sequencing (NGS), is designed to concentrate on chosen genomic segments. By employing polymerase chain reaction (PCR) to selectively amplify these segments for subsequent sequencing, researchers are able to investigate genetic mutations, study microbial variety, and trace evolutionary bonds with remarkable detail and effectiveness.
This method is particularly beneficial when the analytical scope is limited to certain genes or areas, such as the 16S and 18S rRNA genes, ITS regions, and additional genes with functional implications. Its strength in revealing the intricacies of microbial populations has made it a crucial component in the fields of microbiome science, evolutionary biology research, and clinical diagnostics.
Amplicon Sequencing Principle
Amplicon sequencing is an advanced molecular biology technique that employs polymerase chain reaction (PCR) technology to precisely target and replicate specific genomic regions. By designing specialized oligonucleotide primers, researchers can selectively amplify discrete DNA fragments, generating concentrated molecular segments known as amplicons. These amplicons are subsequently analyzed using high-throughput sequencing platforms, enabling researchers to characterize genetic variations with exceptional sensitivity and resolution. The method's remarkable precision allows for comprehensive mapping of genetic elements, making it an invaluable tool for profiling biological systems, detecting molecular mutations, and exploring intricate genomic landscapes across diverse scientific domains. Its ability to provide granular genetic insights with targeted specificity distinguishes amplicon sequencing as a critical technique in contemporary molecular research and diagnostic investigations.
Amplicon Sequencing Workflow
The systematic workflow of amplicon sequencing involves a meticulously structured multi-stage protocol. Initially, researchers extract and preprocess target genetic regions from biological specimens, employing rigorous molecular preparation techniques. The subsequent phase involves PCR, wherein strategically designed primers selectively replicate specific genomic fragments, generating multiple copies of the targeted DNA sequences.
Following amplification, specialized sequencing adapters are methodically ligated to the generated amplicons, constructing a comprehensive genomic library prepared for high-throughput analysis. This crucial step enables the standardization and multiplexing of diverse sample collections, facilitating simultaneous genetic investigation across multiple experimental specimens. By consolidating numerous samples into a unified sequencing pool, researchers can significantly optimize experimental resources, substantially reducing both operational expenses and analytical time requirements.
The integrated approach not only enhances computational efficiency but also provides a robust framework for comprehensive genetic exploration, allowing researchers to extract nuanced molecular insights with remarkable precision and economic effectiveness.
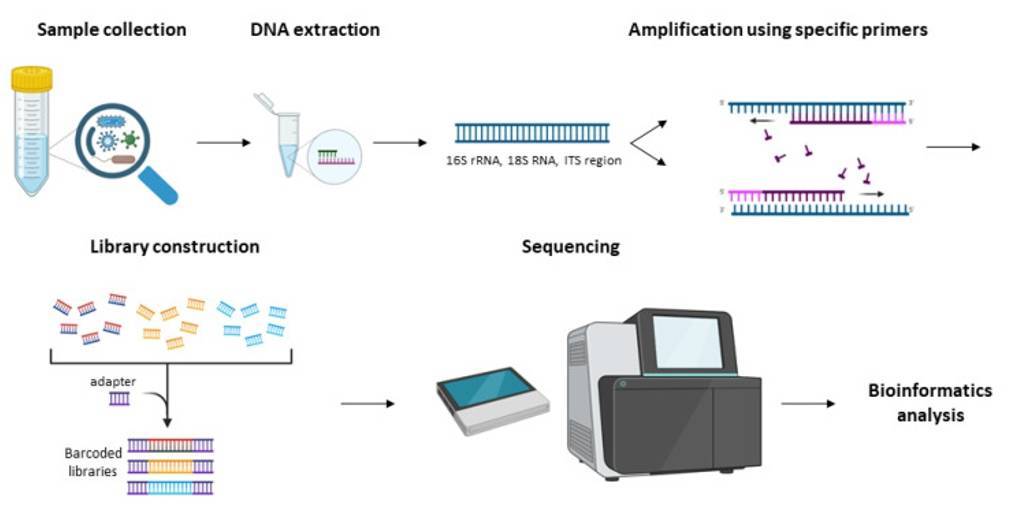 Figure 1. The workflow for the amplicon sequencing(Athanasopoulou, K, et.al, 2023).
Figure 1. The workflow for the amplicon sequencing(Athanasopoulou, K, et.al, 2023).
Amplicon Classification
3.1 16S rDNA
The 16S ribosomal DNA represents a critical molecular marker in prokaryotic taxonomy, encoding a fundamental ribosomal RNA component characterized by intricate genetic domains. This genetic region comprises a complex structure incorporating both conserved and variable genomic segments, rendering it exceptionally valuable for taxonomic classification and phylogenetic investigations.
Characterized by high genomic replication rates and substantial sequence diversity, the 16S rDNA serves as a pivotal molecular tool for microbial identification and systematic exploration of microbial ecosystems. Researchers leverage this genetic region through sophisticated molecular techniques, strategically employing universal primers targeting conserved genomic sequences to selectively amplify specific variable segments.
The methodological approach involves precise PCR amplification, followed by advanced high-throughput sequencing technologies that enable comprehensive molecular characterization. These techniques facilitate detailed taxonomic differentiation, allowing scientists to investigate complex microbial populations across diverse environmental contexts.
Historically significant, this molecular approach was pioneered by Carl Woese, who utilized 16S rDNA sequencing to revolutionize biological classification by introducing the groundbreaking three-domain system. The technique encompasses nine variable and ten conserved genetic segments, providing unprecedented resolution in microbial identification and phylogenetic analysis.By enabling detailed molecular profiling, 16S rDNA sequencing has transformed our understanding of microbial diversity, offering researchers a powerful methodology for exploring the intricate landscapes of microscopic biological systems across multiple ecological domains.
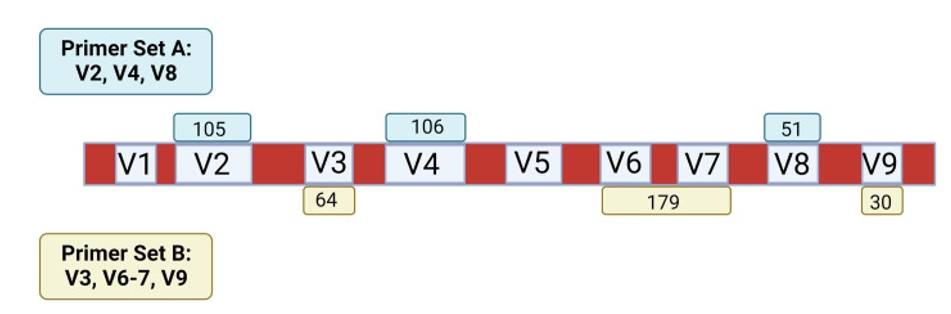 Figure 2. Schematic of the 16S rRNA gene(Maki, K. A, et.al, 2023).
Figure 2. Schematic of the 16S rRNA gene(Maki, K. A, et.al, 2023).
3.2 18S rDNA
18S rDNA encodes the small subunit rRNA of eukaryotic ribosomes. It is more conserved than ITS and is used to study the diversity of eukaryotic microbial communities. Although 18S is effective for constructing phylogenetic relationships, it is less useful for distinguishing species or strains. Combining 18S with ITS, which evolves faster, allows for both detailed evolutionary analysis and improved species resolution, particularly in fungi.
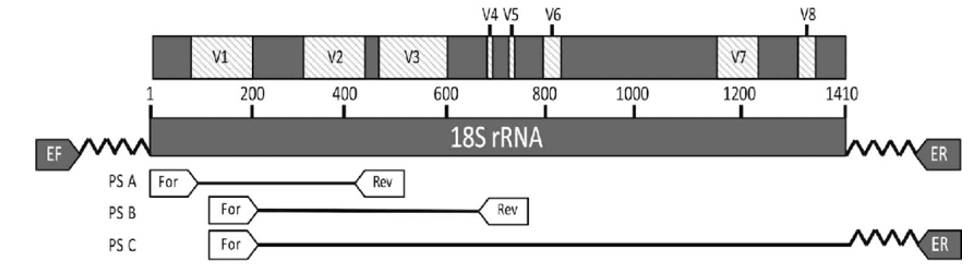 Figure 3 alt. Fish apicomplexan 18S rRNA gene map and diagnostic primers.
Figure 3 alt. Fish apicomplexan 18S rRNA gene map and diagnostic primers.
3.3 ITS
ITS, located between the 18S and 28S rDNA regions, is crucial for identifying fungi, offering a high success rate for species differentiation. However, it is not suitable for distant phylogenetic studies. Combining ITS with 18S sequencing enables both accurate species identification and evolutionary analysis, making it an essential tool for studying fungal community diversity in environmental samples.
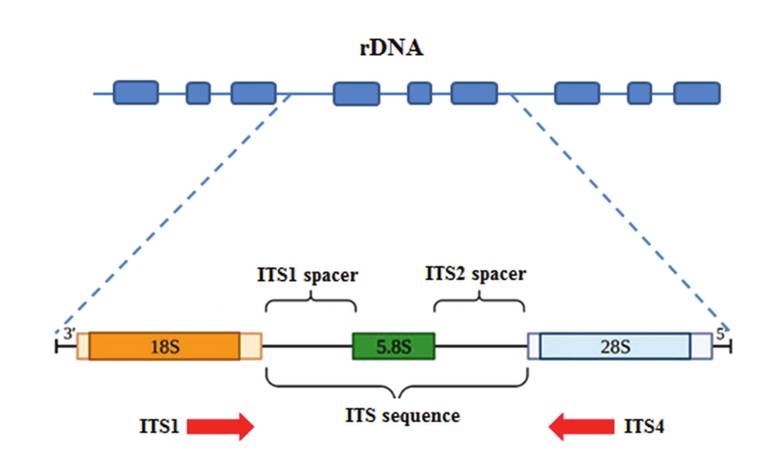 Figure 4. The nuclear ribosomal ITS region.(Ebadi, et.al, 2024).
Figure 4. The nuclear ribosomal ITS region.(Ebadi, et.al, 2024).
Different Sequencing Platforms
The Illumina MiSeq platform is commonly utilized for amplicon sequencing due to its precision and high capacity, which is well-suited for extensive microbial community assessments and 16S/ITS amplicon studies. In contrast, the Ion Torrent PGM system is characterized by rapid and cost-effective sequencing, making it appropriate for quantitative amplicon analyses on a small to moderate scale, despite its shorter read lengths and reduced accuracy. Platforms such as PacBio Sequel II and Oxford Nanopore offer extended read lengths, beneficial for the examination of complete amplicon sequences. However, their increased expenses and the potential for elevated error rates are factors to be taken into account.
| Sequencing Platform |
Platform Type |
Sequencing Method |
Read Length |
Data Accuracy |
Applications |
| Illumina MiSeq |
High-throughput short-read platform |
Bridge amplification library method, paired-end sequencing |
250-300 bp (paired-end) |
High accuracy, highly reliable |
Microbial community diversity analysis, 16S and ITS amplicon sequencing, functional gene analysis |
| Ion Torrent PGM |
Semiconductor sequencing platform |
Semiconductor sequencing technology |
200-400 bp |
Moderate to low accuracy |
Small-scale microbial community analysis, quantitative amplicon sequencing |
| PacBio Sequel II |
Long-read platform |
Single Molecule Real-Time (SMRT) sequencing |
1000-2000 bp |
Moderate but good accuracy |
Long amplicon sequencing, complex microbial genome analysis |
| Oxford Nanopore |
Long-read platform |
Nanopore sequencing |
Variable, average read length 10-50 kb |
Moderate, but higher error rate |
Long amplicon sequencing, microbial community analysis, real-time sequencing |
Applications
The applications of 16S/18S/ITS amplicon sequencing include microbial diversity analysis, environmental monitoring, human microbiome research, food safety, clinical studies, and evolutionary genomics.
1. Microbial Diversity Analysis
One of the most common applications of amplicon sequencing is studying microbial diversity in various environments, including soil, water, and the human body. The approach is particularly effective for:
16S rRNA Sequencing: This technique is widely utilized to profile bacterial and archaeal communities. For instance, a study examining microbial communities in oceanic trench sediments employed 16S rRNA sequencing with novel primers that significantly improved detection of unique microbial groups, revealing 46 bacterial families that were previously undetected using universal primers(Yang, N., et.al, 2022).
18S rRNA Sequencing: This method targets eukaryotic microbes, such as fungi and protists, providing critical insights into their diversity and ecological roles. An example can be seen in research focused on the fungal communities associated with the Cordyceps fungus, where 18S rRNA sequencing revealed a surprisingly high diversity of endogenetic fungal communities(Xia, F. et.al, 2016).
ITS Sequencing: ITS sequencing focuses on fungal communities by leveraging ITS regions as genetic markers for taxonomy. A notable application of this method was conducted in temperate forest soils, where researchers identified 412 sequence types from 863 fungal ITS sequences. This study demonstrated that ITS sequencing could detect greater taxonomic breadth and species richness compared to traditional biotic surveys, thus enhancing our understanding of soil fungal dynamics(O'Brien, H. et.al, 2005).
2. Environmental Monitoring
Amplicon sequencing has emerged as a critical technique in environmental research, allowing investigators to evaluate the microbial makeup across diverse ecological systems, track the influence of contaminants on microbial populations, and study the functions of microbes in biogeochemical processes.
3. Human Microbiome Research
The human microbiome significantly influences health and disease states, and amplicon sequencing has become an essential method for examining the intricacies of this ecosystem. This technique facilitates the detailed analysis of bacterial variation at different anatomical locations, including the intestinal tract, epidermis, and mouth, thereby offering a deeper understanding of the distinctive microbial assemblages within each of these niches.
4. Food Safety and Quality Control
In the realm of food safety and quality assurance, amplicon sequencing serves as a critical technique, offering sophisticated approaches for the identification of pathogens, the surveillance of microbial populations, and the verification of the authenticity of food items.
Clinical and Pathogen Studies
Amplicon sequencing is a crucial technique in clinical studies, providing essential capabilities for the detection and detailed profiling of pathogens that cause infectious illnesses. It is also instrumental in examining genes associated with antibiotic resistance and in exploring the impact of microbial communities on the development of diseases.
6. Evolutionary and Functional Studies
Amplicon sequencing serves as an invaluable asset in the fields of evolutionary and functional genomics, facilitating scientists' insights into the evolutionary connections between species, the identification of genes with functional significance, and the investigation of genetic variation within populations. Through the examination of targeted genetic markers, including 16S rRNA genes or internal transcribed spacer (ITS) regions, researchers are able to construct phylogenetic trees. These trees clarify the evolutionary histories of diverse organisms, providing understanding of the role environmental influences play in the process of biodiversity.
Conclusion
The advent of amplicon sequencing has significantly transformed the methodologies employed by scientists in investigating genetic variability, the complexity of microbial communities, and ecological interactions. This method's precise targeting results in both high specificity and sensitivity, rendering it applicable across a broad spectrum of uses, extending from microbiome profiling to the identification of pathogens. Through the incorporation of techniques like 18S rRNA sequencing, this approach delivers an extensive perspective on the diversity of both eukaryotic and prokaryotic organisms, thereby opening up novel avenues for research in environmental, clinical, and evolutionary biology.
CD Genomics offers advanced amplicon sequencing services, backed by cutting-edge technology and bioinformatics expertise. These services are tailored to meet the unique needs of researchers across various fields, delivering reliable and high-quality data for groundbreaking discoveries.
References
- Athanasopoulou, K., Adamopoulos, P. G., & Scorilas, A. (2023). Unveiling the Human Gastrointestinal Tract Microbiome: The Past, Present, and Future of Metagenomics. Biomedicines, 11(3), 827. https://doi.org/10.3390/biomedicines11030827
- Woese, C. R., Kandler, O., & Wheelis, M. L. (1990). Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proceedings of the National Academy of Sciences of the United States of America, 87(12), 4576–4579. https://doi.org/10.1073/pnas.87.12.4576
- Maki, K. A., Wolff, B., Varuzza, L., Green, S. J., & Barb, J. J. (2023). Multi-amplicon microbiome data analysis pipelines for mixed orientation sequences using QIIME2: Assessing reference database, variable region and pre-processing bias in classification of mock bacterial community samples. PloS one, 18(1), e0280293. https://doi.org/10.1371/journal.pone.0280293
- Renoux, L. P., Dolan, M. C., Cook, C. A., Smit, N. J., & Sikkel, P. C. (2017). Developing an Apicomplexan DNA Barcoding System to Detect Blood Parasites of Small Coral Reef Fishes. The Journal of parasitology, 103(4), 366–376. https://doi.org/10.1645/16-93
- Ebadi, Mostafa & Ebadi, Ali. (2024). Importance and Applications of Endophytic Fungi. https://www.intechopen.com/online-first/1193087.
- Yang, N., Tian, C., Lv, Y., et.al. (2022). Novel primers for 16S rRNA gene-based archaeal and bacterial community analysis in oceanic trench sediments. Applied microbiology and biotechnology, 106(7), 2795–2809. https://doi.org/10.1007/s00253-022-11893-3
- Xia, F., Chen, X., Guo, MY. et al. High-throughput sequencing-based analysis of endogenetic fungal communities inhabiting the Chinese Cordyceps reveals unexpectedly high fungal diversity. Sci Rep 6, 33437 (2016). https://doi.org/10.1038/srep33437
- O'Brien, H. E., Parrent, J. L., Jackson, J. A., Moncalvo, J. M., & Vilgalys, R. (2005). Fungal community analysis by large-scale sequencing of environmental samples. Applied and environmental microbiology, 71(9), 5544–5550. https://doi.org/10.1128/AEM.71.9.5544-5550.2005