
 Sample Submission Guidelines
Sample Submission Guidelines
CD Genomics has been providing the accurate and affordable whole genome sequencing service for couple of years. We combine both Illumina (short reads) and PacBio (long reads) platforms to achieve whole genome de novo assemblies and re-sequencing for viruses, microbes, plants, animals and humans.
The Introduction of Whole Genome Sequencing
Whole genome sequencing represents the determination of the complete DNA sequence of an organism's genome at a single time, which entails chromosomal DNA as well as DNA contained in mitochondria and chloroplasts. Whole genome sequencing provides a powerful tool for both de novo sequencing and re-sequencing. De novo sequencing refers to sequencing of a novel genome without reference sequence available. The coverage quality of de novo assembly depends on the size and continuity of the contigs. De novo sequencing generates the first genome map for a species, thus providing a valuable reference sequence for re-sequencing. Oxford Nanopore and PacBio sequencing systems enable a faster and more accurate characterization of any species at the nucleotide level.
Whole genome resequencing can identify DNA biomarkers such as single nucleotide polymorphism (SNPs), insertions and deletions (indels), structure variations (SVs), copy number variations (CNVs) and other genetic changes of the sequenced species with high accuracy. It also provides an unprecedented opportunity for characterizing the polymorphic variants in a population, which comprehensively unravels the underlying mechanisms of species origin, development, growth, and evolution. What's more, whole genome re-sequencing is an indispensable part of genome-wide association study (GWAS), where common genetic variants in different individuals are assessed to determine if a variant is associated with a particular phenotype. GWAS can be broadly used in food safety, agriculture, and pharmacy, especially personalized medicine.
Advantages of Whole Genome Sequencing
- Single base-pair resolution
- De novo sequencing and genome-wide mutation characterization
- Population evolution and phylogenetic studies
- Disease research, drug discovery and development, and personalized medicine
Whole Genome Sequencing Workflow
CD Genomics combines both Illumina HiSeq and PacBio systems to provide a fast and accurate whole genome sequencing and bioinformatics analysis for any species. Our highly experienced expert team executes quality management, following every procedure to ensure confident and unbiased results. The general workflow for whole genome sequencing is outlined below.

Service Specifications
| Sample Requirements Whole Genome Sequencing:
Note: Sample amounts are listed for reference only. For detailed information, please contact us with your customized requests. |
|
 Click |
Sequencing Strategies
|
 |
Bioinformatics Analysis We provide customized bioinformatics analysis including:
|
Analysis Pipeline
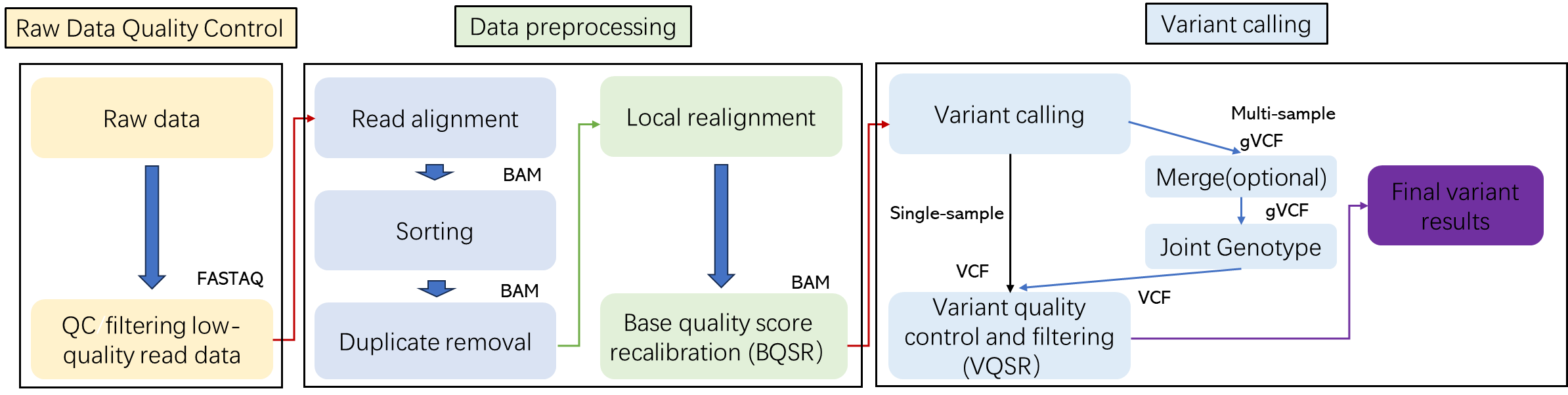
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Whole Genome Sequencing for your writing (customization)
Partial results are shown below:

Distribution of base quality.

Distribution of base content.

Shared SNP number between samples.

SNP mutation type distribution.
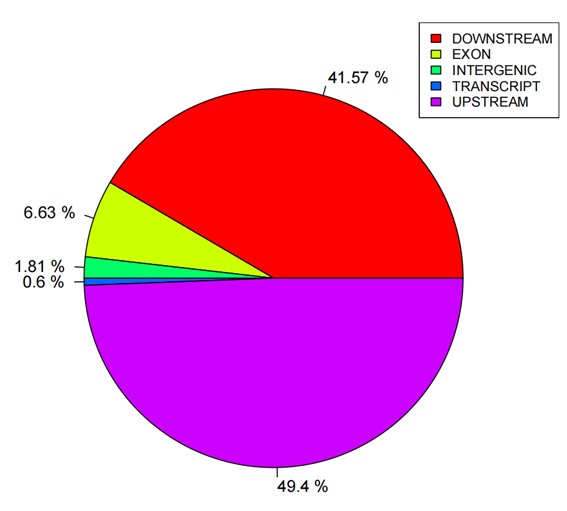
Statistics pie of SNP annotations.

Shared InDel number between samples.

InDel length distribution in both the whole genome scale and CDS regions.

Statistics pie of InDel annotations.
1. How to assure the creditability of assembly results?
Contig N50 and Scaffold N50 are two parameters to describe the "completeness" of a genome assembly. In terms of the quality of assembly results, there are several methods to evaluate it. EST dataset, RNA sequences, or conserved genes can be used to evaluate the completeness of genome assembly. BAC data can be used to test the accuracy.
2. How do we assemble hyper repetitive and heterozygous regions?
Repetitive sequences are abundant in a wide range of species, from microbes to mammals. Repeats have always presented technical challenges for sequence alignment and assembly. As for diploid or polyploid organisms, we generally assemble one set of chromosomes. It is hard to tell which alleles belong to which set of chromosomes in heterozygous regions. We combine the data from HiSeq, PacBio, and Sanger sequencing to ensure the accuracy of genome assembly in hyper repetitive and heterozygous regions.
3. How to estimate the genome size?
There are several ways to estimate the genome size.
i. Websites
For genome size of animals: http://www.genomesize.com/
ii. Flow cytometry
Flow cytometry is a common approach to estimate the genome size.
iii. Genome survey
Genome survey provides an initial view of a genome, and develop strategies for deep sequencing and genome assembly. This method adopts K-mer analysis to estimate genome size, repetition, and heterozygosis mathematically.
4. What are the advantages of whole genome sequencing over whole exome sequencing?
Whole exome sequencing captures only the exons in a genome, while whole genome sequencing capture nucleotides at the genomic level. Given that the exons only account for 1.5% of a genome size, it suggests that whole genome sequencing can capture more sequence data, not just coding regions. Additionally, mutations in noncoding regions may associate with diseases, such as cancer, which leads more people to the studies of noncoding regions.
Whole-genome sequencing identifies rare genotypes in COMP and CHADL associated with high risk of hip osteoarthritis
Journal: Nature Genetics
Impact factor: 27.959
Published: 20 March 2017
Background
Total hip replacement is a treatment for severe hip osteoarthritis, indicated by severe pain and reduced mobility, although radiographic changes may vary. The prior genome-wide association studies (GWAS) of hip osteoarthritis have reported a number of common sequence variants, all conferring small to moderate effects.
Results
In this study, the researchers performed a GWAS to discover the association between end-stage hip osteoarthritis (THR) and sequence variants, based on whole genome sequencing results. The study involved 4,657 Icelandic patients and 207,514 population controls. The authors identified two rare signals that strongly associate with osteoarthritis total hip replacement: (i) a missense variant, c.1141G>C (P.aSP369His), in the COMP gene; (ii) a frameshift mutation, rs532464664 (p.Val330Glyfs*106), in the CHADL gene.
Table 1. Association of c.1141G>C in COMP (0.026%) and the homozygous state of rs532464664 (0.15%) in CHADL with osteoarthritis in Iceland.
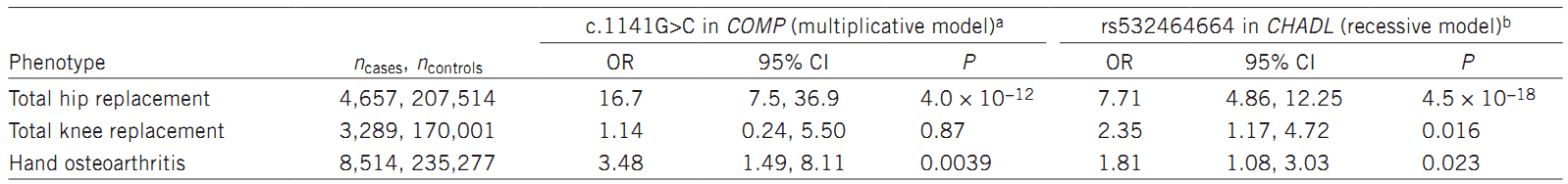
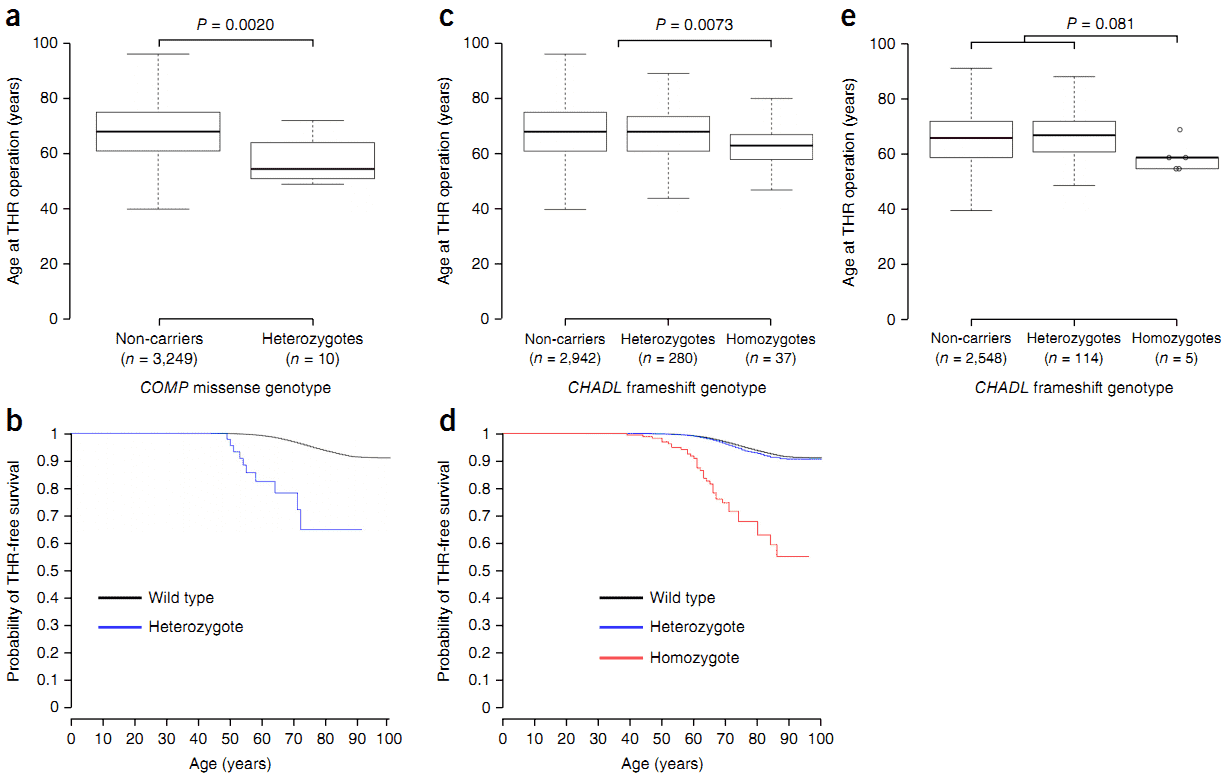 Figure 1. Age at total hip replacement by COMP variant genotype and CHADL variant genotype.
Figure 1. Age at total hip replacement by COMP variant genotype and CHADL variant genotype.
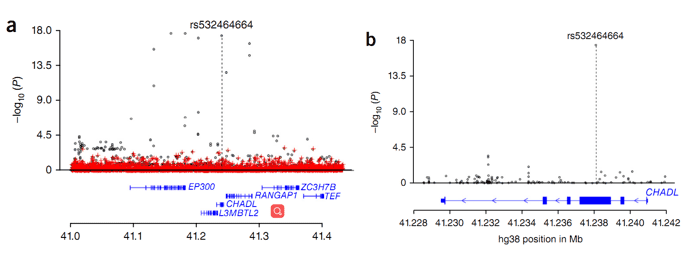 Figure 2. Regional association plot for the CHADL locus.
Figure 2. Regional association plot for the CHADL locus.
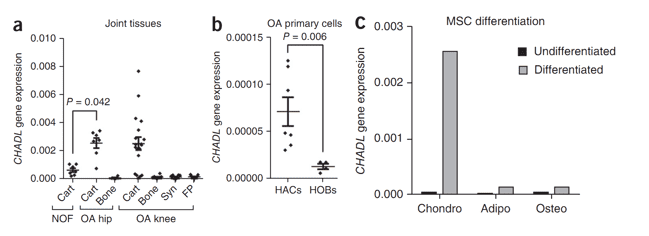 Figure 3. CHADL gene expression in human joint tissues and primary cells and during mesenchymal stem cell differentiation.
Figure 3. CHADL gene expression in human joint tissues and primary cells and during mesenchymal stem cell differentiation.
In summary, the authors identified two variants, including a rare missense variant in COMP and a frameshift mutation in CHADL . They strongly associate with osteoarthritis hip replacement and confer substantial risk of THR (odds ration = 16.7 and 7.7, respectively). Theses odds ratio are higher than those previously reported.
Reference:
- Styrkarsdottir U, et al. Whole-genome sequencing identifies rare genotypes in COMP and CHADL associated with high risk of hip osteoarthritis. Nature genetics, 2017, 49(5): 801.
Here are some publications that have been successfully published using our services or other related services:
Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI
Journal: New phytologist
Year: 2017
High-Density Mapping and Candidate Gene Analysis of Pl18 and Pl20 in Sunflower by Whole-Genome Resequencing
Journal: International Journal of Molecular Sciences
Year: 2020
Isolation and Characterization of Bacteria Associated with Onion and First Report of Onion Diseases Caused by Five Bacterial Pathogens in Texas, U.S.A.
Journal: Plant Disease
Year: 2023
Generation of a highly attenuated strain of Pseudomonas aeruginosa for commercial production of alginate
Journal: Microbial Biotechnology
Year: 2019
Combinations of Bacteriophage Are Efficacious against Multidrug-Resistant Pseudomonas aeruginosa and Enhance Sensitivity to Carbapenem Antibiotics
Journal: Viruses
Year: 2024
Identification of the genetic elements involved in biofilm formation by Salmonella enterica serovar Tennessee using mini-Tn10 mutagenesis and DNA sequencing
Journal: Food Microbiology
Year: 2022
See more articles published by our clients.


