
 Sample Submission Guidelines
Sample Submission Guidelines
The Introduction of GWAS
Genome-Wide Association Studies (GWAS) have revolutionized genetic research by providing a robust methodology for identifying genetic variations correlated with specific traits and diseases. Through comprehensive scanning of the genomes across extensive populations, GWAS seeks to unravel the relationship between genetic variants and phenotypic traits. This methodical approach reveals profound insights into the genetic underpinnings of various conditions, significantly enhancing our comprehension of human genetics and paving the way for the development of targeted therapeutic interventions.
What is the Purpose of GWAS
GWAS are a powerful methodology aimed at identifying genetic variants associated with complex diseases and traits. Unlike traditional family-based linkage studies, GWAS leverages high-throughput genotyping to perform comprehensive scans across the entire genome. This approach enables researchers to detect single nucleotide polymorphisms (SNPs) and other genetic markers correlated with conditions such as diabetes, cardiovascular diseases, and various cancers.
Key Objectives of GWAS
- Identifying Disease-Associated Variants: The primary aim of GWAS is to uncover specific genetic variants that elevate disease risk. For instance, identifying SNPs associated with breast cancer susceptibility enhances our understanding of genetic risk factors and paves the way for targeted therapeutic interventions.
- Understanding Genetic Architecture: GWAS provides insights into how multiple genetic variants interplay to influence complex traits. By elucidating these interactions, researchers can better comprehend the underlying biological mechanisms of diseases and traits, leading to more robust models of genetic architecture.
- Supporting Personalized Medicine: By pinpointing genetic risk factors, GWAS contributes significantly to the advancement of personalized medicine. This precision approach allows for the customization of preventive and therapeutic strategies tailored to individual genetic profiles, thereby improving healthcare outcomes.
Advantages of GWAS
- Comprehensive Coverage: GWAS scan the entire genome, increasing the chances of discovering new genetic markers linked to diseases.
- High Throughput and Scalability: Advanced genotyping technologies allow analysis of many variants across large populations, improving detection power.
- Unbiased Discovery: GWAS use a hypothesis-free approach, reducing bias and uncovering unexpected genetic links to diseases.
- Reproducibility and Validation: Findings from GWAS can be validated in independent studies, ensuring the reliability and accuracy of results.
Applications of GWAS
- Disease Research: Identifying genetic variants linked to diseases such as heart disease and cancer, including SNPs associated with Alzheimer's disease, helps guide future therapeutic developments.
- Pharmacogenomics: Discovering genetic variants that influence individual drug responses enables the creation of personalized drug therapies, enhancing effectiveness and reducing side effects.
- Agricultural Genetics: Finding genetic variants related to beneficial traits in crops and livestock, such as yield and drought resistance, improves agricultural productivity and resilience.
- Population Genetics: Understanding genetic diversity and evolutionary history within populations reveals how variations across ethnic groups affect disease susceptibility.
GWAS Workflow
GWAS involves selecting a study population, genotyping individuals to identify genetic variants, and then using statistical models to find associations between these variants and specific traits or diseases. The results are validated through replication in independent cohorts to confirm their reliability.

Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
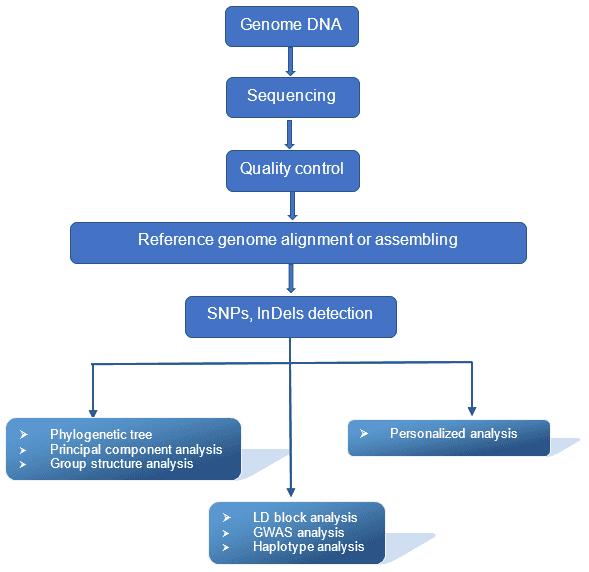
Deliverables
- Raw data (FASTQ)
- Significant SNP information
- QQ-plot and Manhattan plot
- Data analysis report
Partial results are shown below:
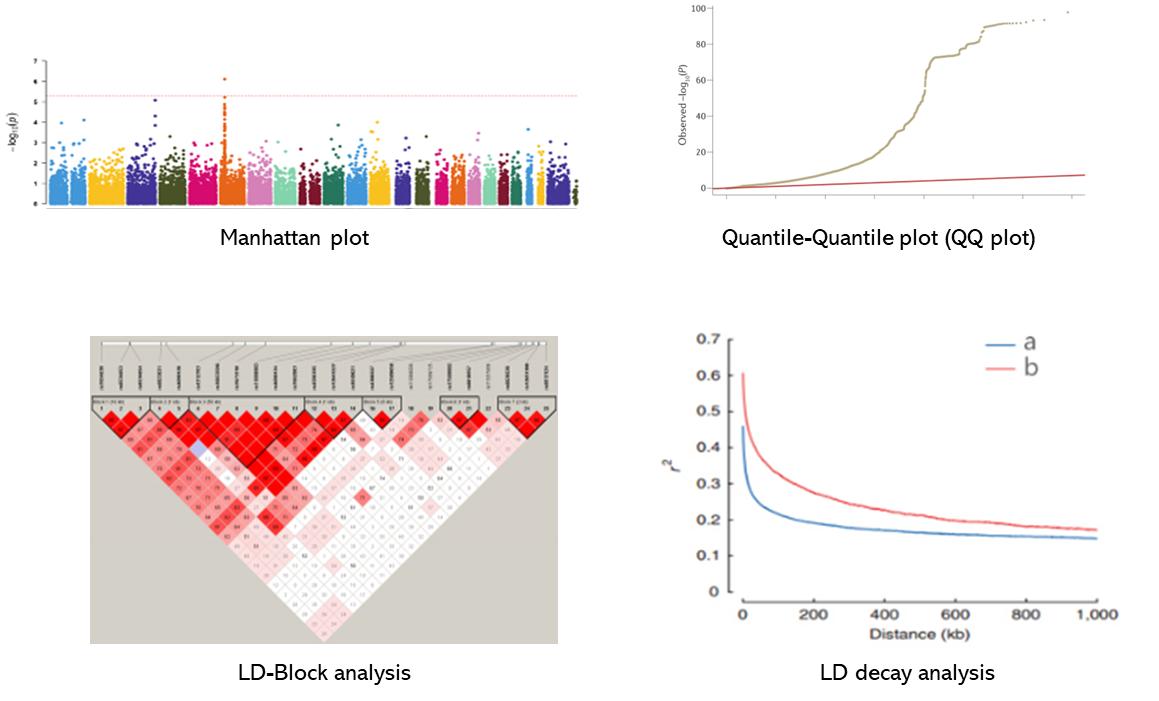
1. What are the principles for sample selection in genome-wide association studies (GWAS)?
- Ensure Sufficient Representativeness of Samples: The selected samples must sufficiently represent the population of interest to ensure that the findings are broadly applicable.
- Avoid Samples with Significant Subpopulation Stratification: Samples should not exhibit marked subpopulation differentiation (e.g., reproductive isolation), as such stratification can introduce substantial genetic background noise, confounding the analysis.
- Focus on Phenotypes with High Heritability: It is advisable to prioritize several key phenotypic traits with high heritability as the primary targets for the study, enhancing the likelihood of detecting significant associations.
- Utilize Binary Traits for Qualitative Characteristics: For qualitative traits, strive to use binary phenotypes (0/1) and ensure that the sample sizes for the two phenotypic categories are approximately equal for robust statistical comparisons.
- Accurately Quantify Quantitative Traits: Quantitative traits should be precisely measured and recorded, such as disease resistance quantified by incidence rate, mortality rate, survival rate, lesion count, or lesion area, rather than using broad categorical scales. The phenotypic data should ideally follow a near-normal distribution.
- Leverage Long-Term Multi-Location Trials for Cultivated Plants: In the case of cultivated plants, conduct multi-year, multi-location, and repeated trials. Results from these trials can be analyzed separately or averaged to strengthen the reliability of association analyses.
- Adjust Sample Size Based on Phenotypic Variability and Control: If phenotypic variation is substantial and controlled by major loci, a smaller sample size (recommended minimum of 200 individuals) may suffice. However, for traits with small phenotypic differences and polygenic control, a larger sample size (recommended minimum of 500 individuals) is necessary to detect significant associations.
2. What are the subjects of GWAS in natural populations?
Non-strictly Genetic Populations:
- Germplasm Resources
- Half-sibling and Mixed Populations
- MAGIC/NAM Populations
- Multiple F2/RIL or Full-Sibling Populations
- Highly Heterozygous Species: F1 Populations
3. Can different traits overlap in a single individual?
Yes, different traits can overlap in the same individual. For example, when categorizing a population based on height and color traits, individuals may be present in both groups. This overlap does not affect the validity of the analysis results.
4. Is GWAS possible without a reference genome?
In the absence of a reference genome, simplified genome sequencing technologies such as RAD-seq or GBS can be employed to detect SNPs through clustering. Although these SNPs can be used for GWAS, the lack of genome annotation limits further gene annotation of identified association loci.
5. How are GWAS results validated?
GWAS results are validated through replication studies in independent cohorts. Additionally, functional studies and pathway analyses help confirm the biological relevance of identified genetic variants.
Genome-wide association and multi-trait analyses characterize the common genetic architecture of heart failure
Journal: Nature Communications
Impact factor: 16.6
Published: 14 November 2022
Background
Heart failure (HF) affects over 38 million people worldwide and is a major cause of cardiovascular issues. Despite its high prevalence, only 11 genetic loci associated with HF have been identified. This study improves GWAS power by combining multi-ancestry data and integrating cardiac imaging traits. It discovers new HF risk variants, identifies relevant tissues, and explores genetic associations with circulating proteins and imaging phenotypes.
Materials & Methods
Sample Preparation
- Human
- Heart failure
- Tissue and cell-type enrichment
Method
- Genome wide association study meta-analysis
- Multivariate GWAS
- Transcriptome-wide association study
- Cross-trait linkage-disequilibrium score regression (LDSC)
- Multi-trait colocalization
- Cardiac gene expression profiling
- Biological pathway and cellular component analysis
Results
A multi-ancestry meta-analysis of HF identified 47 risk loci using data from over 115,000 HF cases and 1.5 million controls. Among these, 939 variants reached genome-wide significance, with 34 loci found beyond previously reported regions. The strongest association was at the PITX2 locus. Replication in additional cohorts confirmed 41 of 44 loci with concordant effects. Pleiotropy scans revealed that many HF loci also associate with other cardiometabolic traits, suggesting shared genetic pathways influencing HF risk.
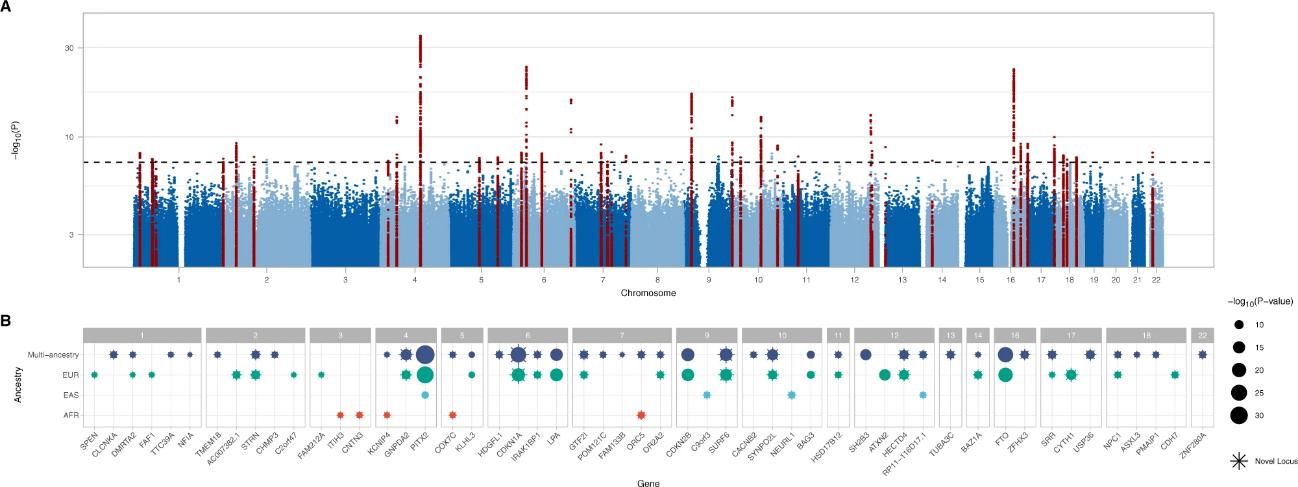 Fig. 1: Genome-wide associations for heart failure.
Fig. 1: Genome-wide associations for heart failure.
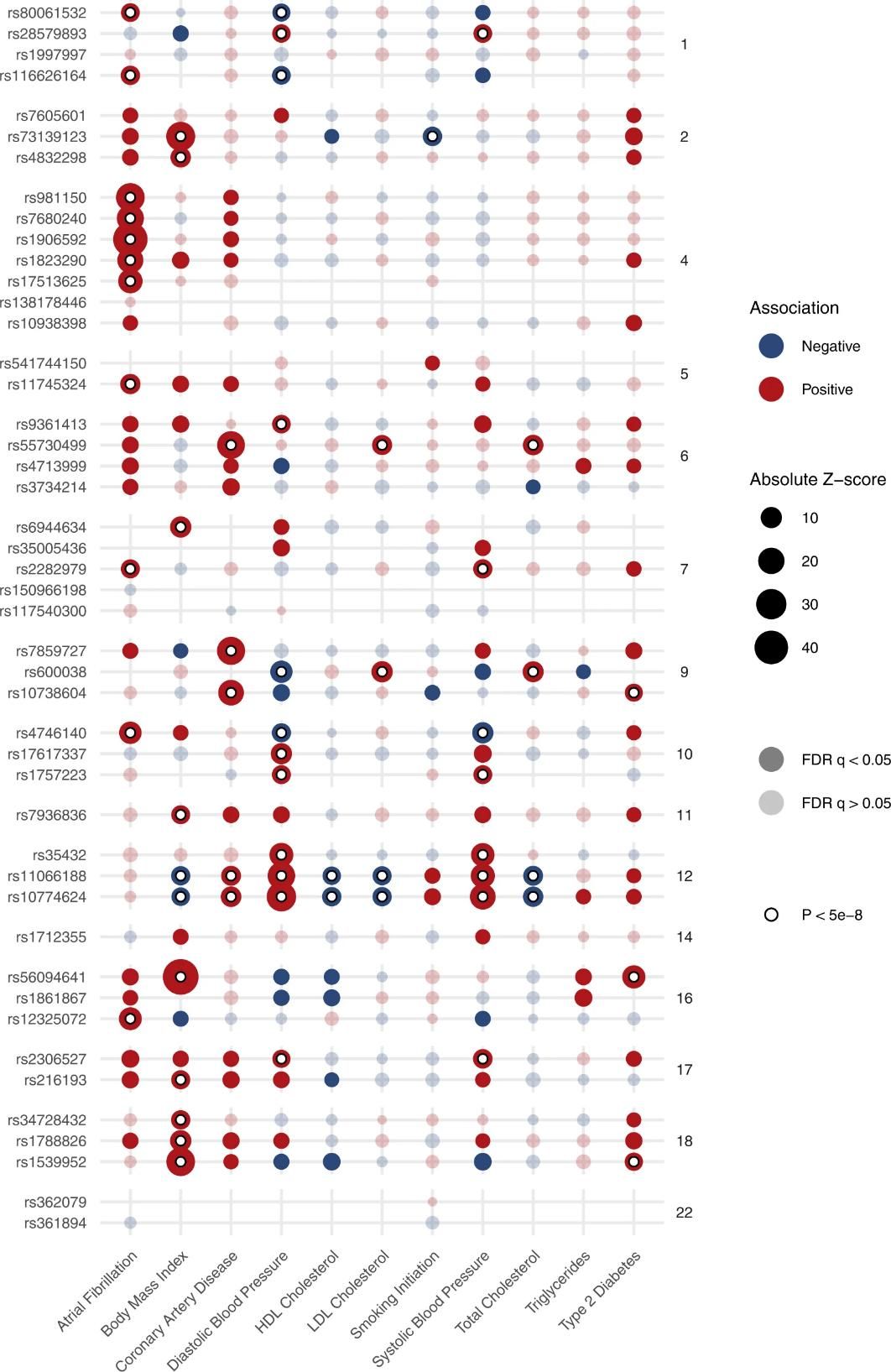 Fig. 2: Associations of heart failure risk variants with common cardiometabolic traits.
Fig. 2: Associations of heart failure risk variants with common cardiometabolic traits.
Using multivariate genetic analysis methods (such as N-GWAMA, MTAG, and Genomic Structural Equation Modeling), researchers identified 61 independent loci associated with HF and related cardiac imaging phenotypes. Among these, 14 were novel discoveries. Many of these loci are enriched near known cardiomyopathy genes, indicating shared genetic etiology with HF and cardiac imaging traits. Colocalization analysis of these loci across multiple traits suggested common genetic causes. Novel associations also linked to other cardiovascular traits and HF risk factors, highlighting the genetic overlap and complexity of HF.
 Fig. 3: Results of multivariate genome wide association study.
Fig. 3: Results of multivariate genome wide association study.
Conclusion
This study used multi-ancestry and multi-trait genome-wide analyses to identify new genetic variants linked to HF and related cardiac traits. It found that integrating different types of genetic data improved the discovery of HF loci, highlighted key genes and pathways involved in HF, and revealed potential links between circulating metabolites and cardiac traits. The findings emphasize the value of combining diverse genetic analyses to better understand HF and its underlying mechanisms.
Reference
- Levin MG, Tsao NL, Singhal P, et. Genome-wide association and multi-trait analyses characterize the common genetic architecture of heart failure. Nature Communications. 2022 Nov 14;13(1):6914.
Here are some publications that have been successfully published using our services or other related services:
Collection of genetic data in ethnic-based studies across Aymaras, Quechuas and Mestizos: the challenges of the Genetics of Alzheimer's in Peruvian Population (GAPP) study
Journal: Alzheimer's & Dementia
Year: 2022
Evaluation of Plasma Biomarkers for A/T/N Classification of Alzheimer Disease Among Adults of Caribbean Hispanic Ethnicity
Journal: JAMA Network Open
Year: 2023
Increased Production of Pathogenic, Airborne Fungal Spores upon Exposure of a Soil Mycobiota to Chlorinated Aromatic Hydrocarbon Pollutants
Journal: Microbiology Spectrum
Year: 2023
A Splice Variant in SLC16A8 Gene Leads to Lactate Transport Deficit in Human iPS Cell-Derived Retinal Pigment Epithelial Cells
Journal: Cells
Year: 2021
See more articles published by our clients.


