
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

CD genomics provides SNP fine mapping service for large SNP number and high volume of samples to help you validate and confirm the SNP loci of interest based on a subset of the detected SNPs.
Single nucleotide polymorphisms (SNPs) represent the most common type of genetic variation among individuals. Fine mapping, an advanced analytical approach, is crucial for pinpointing the specific SNPs that causally influence traits or diseases identified through genome-wide association studies (GWAS).
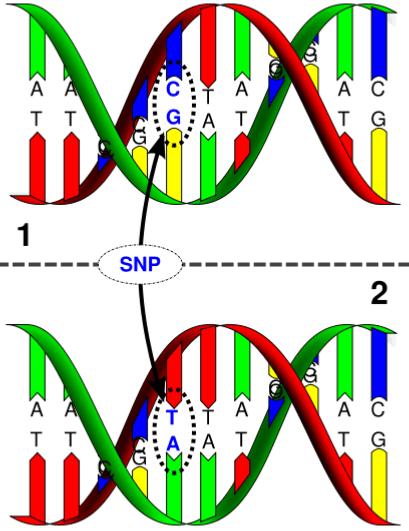
SNP mapping is a fundamental technique in discerning causal variants responsible for genetic associations observed in GWAS. By robustly differentiating between mere statistical associations and genuinely causative SNPs, researchers can elucidate the genetic underpinnings of complex traits and diseases more precisely. For example, while a GWAS might initially flag a broad genomic region associated with a particular disease phenotype, fine mapping refines this resolution to identify specific SNPs that are directly responsible for the observed genetic association. This precision is critical for advancing our understanding of the genetic architecture of diseases.
The process of fine mapping significantly enhances the interpretative power of genetic associations by methodically narrowing down the list of potential causal SNPs within a designated genomic region of interest. This targeted approach facilitates more precise functional studies, enabling researchers to decipher the biological mechanisms through which genetic variants exert their influence on phenotypes. Consequently, fine mapping serves as a pivotal step in translating GWAS findings into actionable biological insights.
Beyond its role in refining genetic associations, fine mapping is instrumental in advancing functional genomics. By uncovering the nuances of how specific genetic variants modulate gene function and regulation, fine mapping provides invaluable insights into the regulatory elements of the genome. Understanding the impact of SNPs on gene expression is crucial for identifying the molecular pathways through which genetic variations contribute to phenotypic outcomes. This detailed knowledge paves the way for the development of targeted therapeutic strategies, emphasizing the importance of fine mapping in the broader context of biomedical research.
Fine mapping of SNPs is a critical step subsequent to large-scale whole genome SNP genotyping studies. It serves to hone in on specific genes that are potentially associated with the phenotypic traits of interest. Typically, fine mapping endeavors involve a reduced number of SNPs but utilize a substantially larger sample size to increase the accuracy of the association.
Upon completion of a comprehensive genome-wide SNP screening and the identification of preliminary target regions, fine mapping is imperative. An effective genotyping platform for fine mapping must demonstrate a high call rate for the chosen SNPs, operate at a relatively high multiplex level, and avoid protracted assay optimization processes.
Platforms based on primer extension and allele-specific ligation have proven efficacious for fine mapping applications. These methods are well-suited to achieving the necessary precision and efficiency required for identifying causal variants that contribute to phenotypic variation and disease susceptibility.
Heuristic approaches constitute some of the earliest techniques employed in SNP fine mapping. These methods rely on empirical and intuitive judgments to filter SNPs based on their pairwise correlation with a lead SNP. SNPs displaying high correlation with the lead SNP are considered potential causal variants. However, these methods are often constrained by arbitrary thresholds and lack an objective measure of causality.
Penalized regression models offer solutions to the issues of instability and overfitting commonly encountered in high-dimensional data analysis. By introducing penalties during the likelihood maximization process, these models facilitate the selection of SNPs that exhibit a strong association with the trait while minimizing biases from smaller effect estimates. Well-known models in this category include Lasso, Elastic Net, and Minimax Concave Penalty.
Bayesian methods provide a more sophisticated framework by calculating posterior probabilities for SNPs having non-zero effects. This approach yields posterior inclusion probabilities (PIP), which quantify the evidence supporting each SNP as a causal variant. Bayesian methods often employ Markov Chain Monte Carlo (MCMC) techniques to approximate integrals, thereby enhancing resolution through model comparison and the integration of functional annotation data.
Integrating genome annotation data enhances fine mapping models by incorporating gene regulatory and expression quantitative trait loci (eQTL) information. This integrative approach refines causal SNP identification by weighting them according to their biological relevance and adjusting prior probabilities within Bayesian models, thus improving the overall resolution.
Trans-ethnic fine mapping exploits genetic diversity across different populations to increase resolution. By combining GWAS results from various ethnic groups, this approach capitalizes on differences in linkage disequilibrium (LD) patterns to more precisely identify the loci of causal SNPs. This method proves especially valuable in genomic regions exhibiting varied genetic structures across populations.
We offer several assay types for verifying SNP markers discovered by RADseq, GBS, SNP chips or similar technologies. They include:
MassARRAY SNP Genotyping employs MALDI-TOF MS to detect SNPs by analyzing mass differences of extended DNA products. This technique offers high sensitivity, accuracy, and cost-effectiveness. It is particularly advantageous for high-throughput genotyping, though it requires high-quality samples and can be sensitive to complex sequences around the SNP sites.
SNaPshot SNP Genotyping uses fluorescently labeled primers and capillary electrophoresis to analyze SNPs. It allows multiplexing of multiple SNPs in a single reaction, making it suitable for medium-throughput projects. While it requires specialized reagents and can be more costly, it provides high flexibility and robustness in detecting diverse SNPs.
TaqMan SNP Genotyping utilizes real-time PCR with fluorescently labeled probes to precisely identify SNPs based on emitted fluorescence. This method is highly specific and reliable, ideal for large-scale studies. It provides excellent accuracy and is widely recognized for its ease of use, though it involves probe synthesis and can be relatively expensive.
Genotyping platforms employed in SNP fine mapping are capable of both uniplex and multiplex genotyping. Notable uniplex platforms include the TaqMan SNP Genotyping and SNaPshot, while the MassARRAY SNP Genotyping represents a reliable option for multiplex genotyping. These platforms are distinguished by their high degree of flexibility and adaptability. Users can balance throughput by adjusting the number of SNPs analyzed in conjunction with the sample size, according to specific investigational needs. Additionally, they offer a pragmatic advantage in that failed SNP assays can be swiftly redesigned and reordered, thereby enhancing the efficiency and accuracy of genetic analyses.
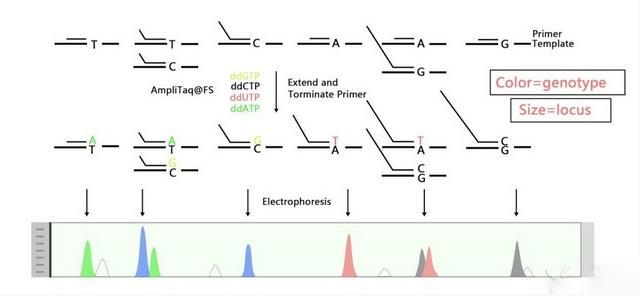 Figure 1. The principle diagram of SNaPshot.
Figure 1. The principle diagram of SNaPshot.
Our team of experts provides consultation services to assist you in determining the optimal genotyping strategy and experimental design tailored to meet your specific research objectives in a cost-effective and timely manner.
Embryonic origin and genetic basis of cave associated phenotypes in the isopod crustacean Asellus aquaticus
Scientific Reports | 2018Scanning indels in the 5q22.1 region and identification of the TMEM232 susceptibility gene that is associated with atopic dermatitis in the Chinese Han population
Gene | 2017Investigating interactive effects of worry and the catechol-o-methyltransferase gene (COMT) on working memory performance
Cognitive, Affective, & Behavioral Neuroscience | 2021Impact of the OATP1B1 c.521T>C single nucleotide polymorphism on the pharmacokinetics of exemestane in healthy post-menopausal female volunteers
Journal of clinical pharmacy and therapeutics | 2017

