
 Sample Submission Guidelines
Sample Submission Guidelines
RAD-seq offers a cost-effective way to uncover genetic variations and build genetic maps. CD Genomics provides advanced RAD-seq techniques, including dd-RAD and 2b-RAD, to deliver high-quality genomic insights tailored to your research needs.
What is RAD-Seq
Restriction-site-associated DNA sequencing (RAD-seq) is a robust method for analyzing genomic DNA at sites recognized by specific restriction endonucleases. Originally introduced by Baird et al. in 2008, the traditional RAD-seq approach involves a single restriction enzyme digestion of genomic DNA, followed by the addition of adaptors and subsequent random shearing of these fragments via sonication. This methodology primarily yields read 1 sequences that align precisely with the enzyme's recognition sites, whereas read 2 sequences exhibit considerable variability due to random fragmentation.
RAD-seq offers substantial advantages in studies involving species without a reference genome. One of its primary benefits is the significant reduction in sequencing costs compared to whole-genome sequencing, all the while providing a wealth of genome-wide variation data. These attributes make RAD-seq particularly valuable in diverse applications, such as molecular marker development, genetic map construction, gene and quantitative trait loci (QTL) mapping, genome-wide association studies (GWAS), population genetics, and molecular breeding.
Variations of RAD-seq, including double digest-RAD (dd-RAD), 2b-RAD, and specific locus amplified fragment-RAD (SLAF-RAD), have been developed to address different experimental needs. The differences among these techniques primarily involve the number of enzymes used, the presence or absence of random fragmentation steps, and the design of adaptors with or without barcodes. Despite these variations, the core principle remains consistent: all involve digesting the genome with restriction enzymes and sequencing the resultant fragments.
- dd-RAD: This method employs a combination of rare and frequent cutting restriction enzymes to digest the genome, eliminating the need for a subsequent fragmentation step. It allows more precise control over fragment size and yields reproducible results.
- 2b-RAD: Utilizes type IIB restriction enzymes, which produce DNA fragments of uniform size. This uniformity simplifies downstream processes and enhances the consistency and comparability of results.
- SLAF-RAD: Shares similarities with dd-RAD in the digestion step but employs non-specific adaptors during library preparation. This approach aims to balance specificity with flexibility in different experimental setups.
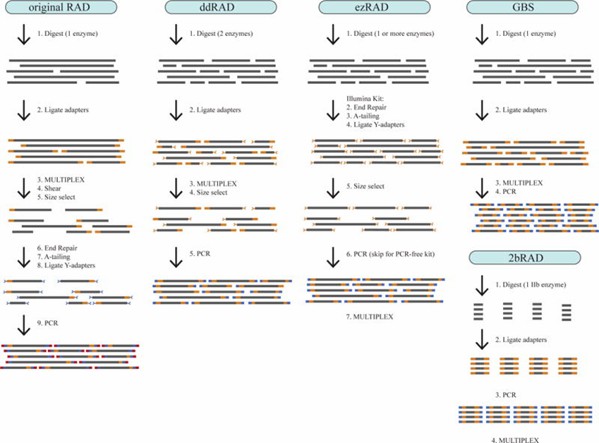 Figure 1. Step-by-step illustration of five RAD-seq library prep protocols. (Andrews et al., 2016)
Figure 1. Step-by-step illustration of five RAD-seq library prep protocols. (Andrews et al., 2016)
What is RAD-Seq Used for
RAD-seq is a versatile tool employed across a range of genomic applications:
- Molecular Marker Development: Utilized to discover genetic markers essential for breeding programs and genetic research.
- Genetic Mapping: Facilitates the creation of high-density genetic linkage maps, enabling detailed analysis of genetic linkage and inheritance patterns.
- Gene/QTL Mapping: Aids in pinpointing QTL and specific genes associated with particular traits.
- GWAS: Assists in identifying genetic variants linked to complex traits or diseases within populations.
- Population Genetics Analysis: Enables the examination of genetic diversity, population structure, and evolutionary relationships among individuals and species.
Advantages of Our RAD-Seq Service
- Provides total genetic distance of the map and is responsible for trait localization for clients.
- Provides clients with text for the methods section of their articles, as well as project-related tables and figures.
- Flexible pricing strategy.
- Extensive experience in analyzing large animal and plant genomes.
- Integration of comprehensive genomics analysis, offering various omics technology services, including genomics, transcriptomics, and epigenetics, as well as advanced and customized genomics analysis solutions.
- Streamlined processes with shorter project cycles.
- Cost-saving through joint analysis of sample information with public databases.
RAD-Seq Workflow
RAD-seq entails a structured series of stages, including:
- Genomic DNA fragmentation employing restriction endonucleases.
- Adjoining P1 adapters to the fragmented DNA.
- Aggregating and randomly fragmenting enzyme-cleaved segments from all samples.
- Culling fragments of appropriate dimensions (typically in the range of 300 to 700 base pairs).
- Ligating P2 adapters to the selected fragments.
- Implementing PCR amplification for enrichment.
The workflow of RAD-Seq at CD Genomics involves several key steps:

Service Specifications
Our service specifications mainly encompass three aspects: Sample Requirements, Sequencing Strategy, and Bioinformatics Analysis.
Sample Requirements
1. Variant Detection:
DNA sample quantity: ≥ 3 µg.
Digestion with EcoRI (GAATTC).
Recommended sequencing depth: ≥ 1X per sample (≥ 5X for assembly samples).
2. Genetic Mapping:
DNA sample quantity: ≥ 3 µg.
Sequencing depth: Parents 2-5X, Offspring 0.8X per individual (F1, F2, etc., temporary populations), 0.6X per individual (RIL, DH, etc., permanent populations).
Applicable to: Haploid or diploid species, all mapping populations (F1, F2; RIL, DH, etc.), with populations of 100 individuals or more.
DNA sample quantity: ≥ 3 µg.
Sequencing depth: ≥ 1X per individual (≥ 5X for assembly samples).
Applicable to: Different subpopulations within haploid or diploid species, distinct subpopulation divisions, representative individuals within the same subpopulation. Approximately 10 samples per subpopulation (for animals ≥ 10, for plants ≥ 15), with a total of at least 30 samples.
4. Sample Details:
DNA samples: Please provide DNA with a concentration > 100 ng/μl and a total amount > 3 μg, OD 260/280 ratio between 1.8 and 2.0.
Ensure that the DNA is not degraded, shows no significant RNA bands upon electrophoresis, and has clear, intact genomic bands, with the main band above 100 kb. Contamination by polysaccharides or glycoproteins can pose significant challenges to DNA fragmentation, making removal difficult. Therefore, it is essential that provided samples are free from polysaccharide or glycoprotein contamination.
Plant samples: Select young plant tissues, with each sample weighing > 500 mg. Store and ship the samples on dry ice or in liquid nitrogen.
Animal samples: Fresh animal tissues are required, avoiding fatty tissues, with each sample weighing > 50 mg.
For common species, select tissues such as liver, kidney, and blood. For rare species, provide ear samples or hair samples (with hair roots) with lower fat content. To minimize the impact of individual variation on subsequent assembly, it is preferable to sample from the same individual. If the species are small in size and the DNA extraction quantity from one individual is insufficient for sequencing, try to reduce the number of sampled individuals while maintaining the required amount. Provide tissue samples of > 50 mg; provide a sufficient quantity, considering different species may yield varying amounts of DNA upon extraction.
Note: Sample amounts are listed for reference only. For detailed information, please contact us with your customized requests.
Sequencing Strategy
| Product Name | Sequencing Platform | Parameter Specifications |
|---|---|---|
| RAD-Seq-Variation Detection | HiSeq PE150 | ≥1X per individual (≥5X for assembly samples) |
| RAD-Seq-Genetic Mapping | HiSeq PE150 | Parent: 2-5X, Offspring: 0.8X per individual (F1, F2); 0.6X per individual (RIL, DH) |
| RAD-Seq-Population Evolution | HiSeq PE150 | ≥1X per individual (5X for assembly samples) |
- Molecular Marker Development: Identification of individual SNPs, identification of individual InDel markers, integration of population SNP markers.
- High-Density Genetic Mapping: Differentiation of linkage groups, ordering of large numbers of markers, and screening of biased segregation loci.
- Population Genetics Analysis: Conducting population genetic analyses of SNP data, including tree construction, population structure analysis, and PCA analysis.
- Genomic Sketch Mapping: Assembly of genomic sketches and gene annotation.
Note: Recommended data outputs and analysis contents displayed are for reference only. For detailed information, please contact us with your customized requests.
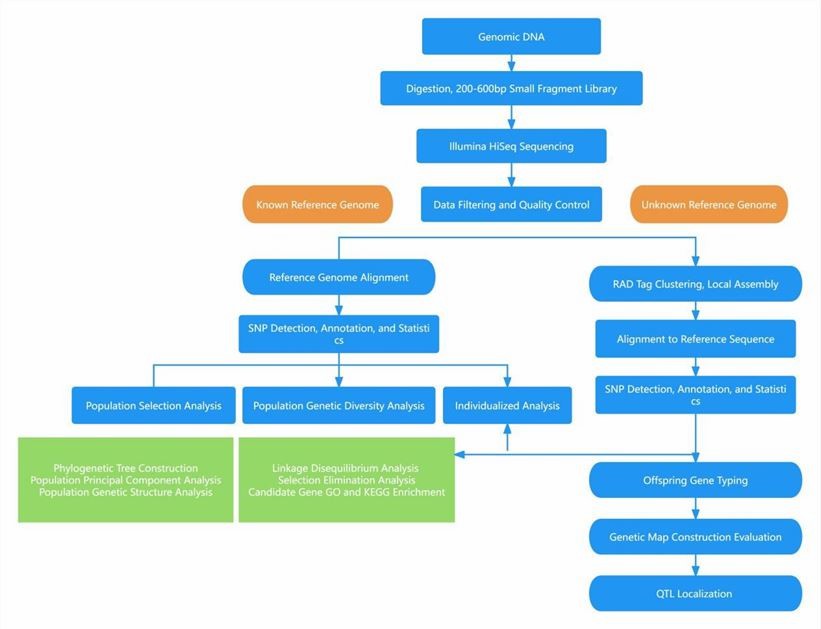
Analysis Pipeline
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in RAD-seq for your writing (customization)
Reference
- Andrews KR, Good JM, Miller MR, et al. Harnessing the power of RADseq for ecological and evolutionary genomics. Nature Reviews Genetics. 2016 Feb;17(2):81-92.
Partial results are shown below:
Q: What considerations are paramount in the development of high-density SNP markers?
A: The essential prerequisites and guiding principles for the development of high-density SNP markers revolve around the imperative of achieving a balanced distribution of fragments throughout the genome. By identifying sequences that faithfully represent comprehensive genomic information, an array of thousands of SNP markers can be systematically generated. In the initial stages, bioinformatics techniques are deployed to methodically assess the reference genome of the target species (or known BAC sequences). Guided by factors such as genome GC content, repetitive sequence characteristics, and gene attributes, discerning choices of appropriate restriction enzymes and sequencing library types are made, with the primary objective of ensuring that the density, consistency, and overall efficacy of molecular markers align with the stringent requisites of genetic analysis and molecular breeding.
Q: What are the application domains of RAD-seq?
A: RAD-seq finds versatile utility in diverse research areas encompassing the establishment of genetic maps, phylogeographic analysis of polymorphisms, association studies, and QTL mapping, irrespective of the presence or absence of reference genome data for a given species. The broad reach of RAD-Seq technology extends to investigations involving variation discovery, the construction of genetic maps, the exploration of functional genes, and the study of population evolution.
Q: What are the merits of employing RAD-seq in the context of population evolution studies?
A: Population evolution studies typically encompass a considerable number of specimens. RAD-seq presents a set of distinct advantages in this regard. It effectively diminishes genome complexity, resulting in reduced expenditures on library preparation and sequencing. Its straightforward workflow streamlines the process. Furthermore, its independence from reference genomes renders it applicable to a broader spectrum of species. Notably, RAD-Seq excels in investigations involving extensive sample sizes, thereby furnishing a robust framework for comprehensive data extraction through whole-genome resequencing technologies.
Q: What distinguishes the utilization of a reference genome from its absence?
A: When it comes to the identification of mutational information, the incorporation of a reference genome for sequence alignment and variation detection typically yields heightened levels of both efficiency and precision. Additionally, it facilitates the precise localization of specific genes and enables investigations into population linkage disequilibrium.
Genome sequence and analysis of the Japanese morning glory Ipomoea nil
Journal: Nature Communications
Impact factor: 16.6
Published: 08 November 2016
Background
Ipomoea is a large genus with diverse species, including the commercially significant I. nil (Japanese morning glory). Researchers used Illumina and PacBio sequencing to assemble a 750 Mb genome of I. nil, identifying Tpn1 transposons and mapping the dwarfism gene, CONTRACTED.
Materials & Methods
Sample Preparation
- Morning glory
- DNA extraction
Sequencing
- Whole genome sequencing
- mRNA-Seq
- RAD-seq
- Genome assembly
- Linkage map construction
- Repeat analysis
- Gene prediction
- Comparative analysis
Results
1. DNA Sequencing and Genome Assembly
A single TKS plant was sequenced, revealing a 750 Mb genome. PacBio sequencing provided long reads and high coverage, which, combined with Illumina data, produced an assembly with an N50 of 3.72 Mb. An alternative Illumina-only assembly was larger but of lower quality.
2. Mis-assembly Detection and Pseudo-molecule Construction
RAD-seq data helped correct mis-assemblies, leading to an improved assembly. Pseudo-chromosomes were constructed, covering 91.42% of the genome with an N50 of 44.78 Mb.
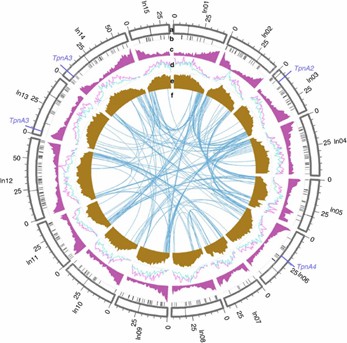 Figure 1: Genomic characterizations of I. nil.
Figure 1: Genomic characterizations of I. nil.
3. Assembly Validation
The assembly was validated through CEGMA and BUSCO analysis, showing high completeness, and supported by alignments with ESTs and BAC-end sequences, confirming its quality.
4. Repeat Analysis and Identification of Tpn1 Transposons
The genome contained 63.29% repetitive elements, including Tpn1 transposons, some of which were active and linked to gene disruptions in mutants.
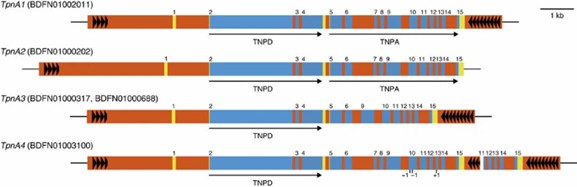 Figure 2: The Tpn1 family transposons encoding transposases.
Figure 2: The Tpn1 family transposons encoding transposases.
5. Gene Prediction and Functional Annotation
42,783 gene models were predicted, with 79.12% annotated, and RNA-seq data provided functional insights.
6. Analysis of the Dwarf Gene CONTRACTED (CT)
The CT gene, associated with dwarfism and BR biosynthesis, was disrupted by transposon insertions in mutants, affecting gene expression.
7. Genome Evolution
Phylogenetic analysis revealed I. nil's close relationships with tomato and kiwifruit, with unique gene families and WGD events distinct from Solanaceae.
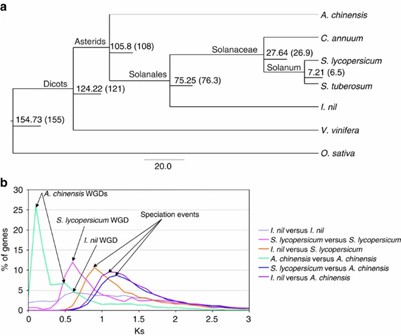 Figure 3: Genome evolution.
Figure 3: Genome evolution.
Conclusion
The study utilized advanced sequencing tools to produce a high-quality genome assembly of Ipomoea nil, significantly improving scaffold lengths and repeat resolution. It identified key genes and transposons related to plant traits and mutations, and highlighted the impact of whole-genome duplications on plant evolution. This work supports future research and comparative genomics in the Solanales order.
Reference
- Hoshino A, Jayakumar V, Nitasaka E, et. Genome sequence and analysis of the Japanese morning glory Ipomoea nil. Nature communications. 2016 Nov 8;7(1):13295.
Here are some publications that have been successfully published using our services or other related services:
Use of biostimulants for water stress mitigation in two durum wheat (Triticum durum Desf.) genotypes with different drought tolerance
Journal: Plant Stress
Year: 2024
The Restriction-Modification Systems of Clostridium carboxidivorans P7
Journal: Microorganisms
Year: 2023
In the land of the blind: Exceptional subterranean speciation of cryptic troglobitic spiders of the genus Tegenaria (Araneae: Agelenidae) in Israel
Journal: Molecular Phylogenetics and Evolution
Year: 2023
Genetic Modifiers of Oral Nicotine Consumption in Chrna5 Null Mutant Mice
Journal: Front. Psychiatry
Year: 2021
A high-density genetic linkage map and QTL identification for growth traits in dusky kob (Argyrosomus japonicus)
Journal: Aquaculture
Year: 2024
Genomic and chemical evidence for local adaptation in resistance to different herbivores in Datura stramonium
Journal: Evolution
Year: 2020
See more articles published by our clients.


