What are the Technical Advances in RNA-seq
Sanger sequencing and microarrays. Sanger sequencing technology was first used for transcriptomics, which enabled methods such as SAGE (serial analysis of gene expression). SAGE was one of the first attempts to quantify gene expression on a global basis. Almost instantaneously, microarrays utilizing complementary probe hybridization, quickly emerged and come to dominate the field of transcriptomics profiling for the next decade.
NGS. The advent of next-generation technologies has enabled the sequencing approach to surpass microarray approach. In 2006, the first RNA-seq paper was published by utilizing454/Roche technology. The era of RNA-seq dominance began in 2008 with the maturity of Illumina/Solexa technology. The most popular technical platforms for RNA-Seq has been the Illumina Genome Analyzer and Hi-Seq. While the Illumina/Solexa technology can generate gigabases of data per run (initially 1GB per run for the Genome Analyzer in 2006 and 600 GB per run for the HiSeq in 2012), Roche/454 technology generates reads long enough for RNA-seq but are hampered by the relatively low throughput and high cost.
Third generation sequencing. Despite the popularization of the NGS technologies, the application of third generation sequencing in RNA-seq is on its way. For example, Heliscope sequencing and single-molecule real-time (SMRT) sequencing have already been applied in some RNA-seq studies. PacBio SMRT long reads sequencing technology can easily cover complete transcript from the 5' end to the 3'-poly A tail without the need of fragmentation to obtain full-length cDNA sequences, which is useful to identify new transcripts and new introns, thereby accurately identifying isoforms, alternative splicing sites, fusion gene expression, and allelic expression.
Services you may interested in
Table 1. The advantages of RNA-seq compared with other transcriptomics approaches (Wang et al. 2009).
| Technology |
Tiling microarray |
cDNA or EST sequencing |
RNA-seq |
| Technology specifications |
| Principle |
hybridization |
Sanger sequencing |
High-throughput sequencing |
| Resolution |
From several to 100 bp |
Single base |
Single base |
| Throughput |
High |
Low |
High |
| Reliance on genomic sequence |
Yes |
No |
In some cases |
| Background noise |
High |
Low |
Low |
| Application |
| Simultaneously map transcribed regions and gene expression |
Yes |
Limited for gene expression |
Yes |
| Dynamic range to quantify gene expression level |
Up to a few-hundredfold |
Not practical |
>8,000-fold |
| Ability to distinguish different isoforms |
Limited |
Yes |
Yes |
| Ability to distinguish allelic expression |
Limited |
Yes |
Yes |
| Practical issues |
| Required amount of RNA |
High |
High |
Low |
| Cost for mapping transcriptomes of large genomes |
High |
High |
Relatively low |
What are the Challenges of RNA-seq
- Short-read. Illumina sequencing technology has steadily increased read length and throughput since its introduction in 2007. Long paired-end strand-specific reads are commonly used for higher levels of mappability and de novo assembly of transcriptomes. Furthermore, the third generation sequencing technology (such as PacBio and Ion-Torrent) enables full-length transcripts sequencing.
- PCR biases. Another concern is the impact of PCR amplification on the accuracy of gene expression quantitation via RNA-seq. Helicos and some of the third sequencer used an amplification-free technology. There are also PCR-free methods for Illumina sequencing.
Workflow of RNA-seq based on NGS
The workflow of RNA-seq by utilizing high-throughput sequencing technology is illustrated in Figure 1. Briefly, long RNAs are first converted into a library of cDNA fragments through RNA or DNA fragmentation. Sequencing adaptors are then attached to each cDNA fragment and sequence data are generated in a high-throughput manner from both ends (paired-end sequencing). The resulting sequence reads are subsequently aligned with the reference genome or transcriptome, and are classifies into three types: exonic reads, junction reads and poly(A) end-reads. A base-resolution expression profile can be generated by using these three types of sequence reads.
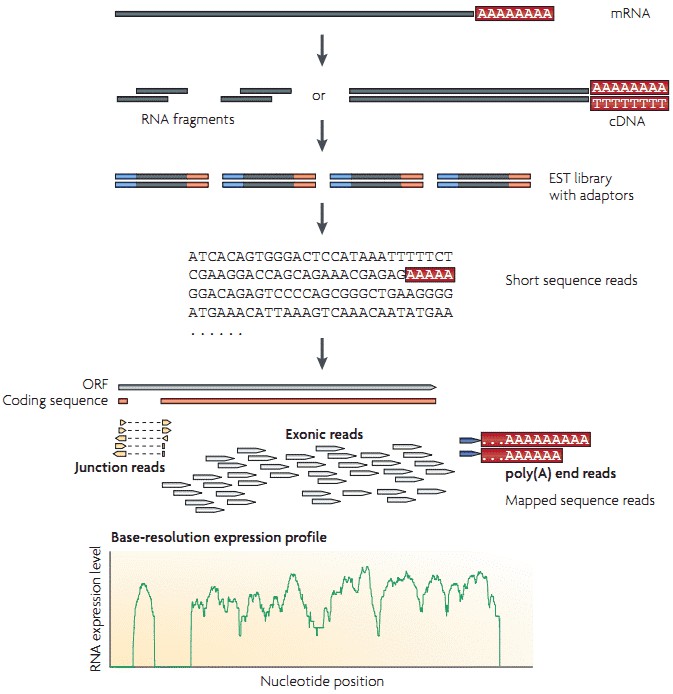 Figure 1. A typical workflow of RNA-seq (Wang et al. 2009).
Figure 1. A typical workflow of RNA-seq (Wang et al. 2009).
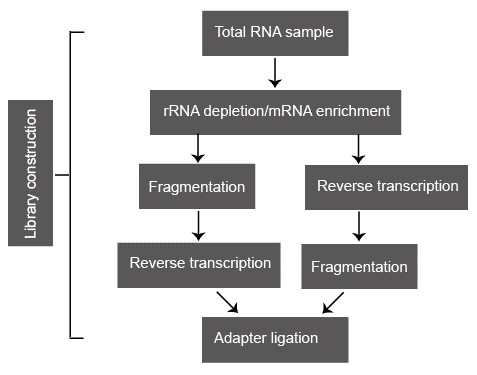 Figure 2. A typical library construction pipeline of RNA-seq.
Figure 2. A typical library construction pipeline of RNA-seq.
Following sample collection, total RNA is usually isolated via organic extraction and/or silica-membranes of spin columns. Total RNA sample is subsequently processed either by direct selection of poly(A) RNA or by selective removal of rRNA because the abundant rRNA is usually not the research focus and greatly reduces the coverage of the useful transcript. Oligo(dT)-based mRNA purification procedure is widely used in eukaryotes. However, some RNA transcripts that lack the poly(A) tails are missed. Compared to the poly(A) RNA selection, ribo-depletion approach is preferred because it enriches all nonribosomal RNA species, including tRNA, ncRNAs, nonpoly(A) mRNA, and preprocessed RNA. The two most popular rRNA depletion methods are: (i) hybridization of rRNA with biotin-labeled anti-rRNA probes, followed by removal with streptavidin-coated magnetic beads; and (ii) selective degradation of rRNA by a 5'-3' exonuclease that specifically recognizes rRNA with a 5' phosphate.
Fragmentation is subsequently conducted to reach the desired length for different NGS technologies. Some small RNAs, such as microRNAs, piwi-interacting RNAs, and short interfering RNAs, can be directly sequenced without fragmentation. Larger RNA molecules need to be fragmented into smaller pieces (200-500 bp) before deep-sequencing technologies. cDNA fragmentation (DNase I treatment or sonication) and RNA hydrolysis or nebulization. However, each of these methods can create a different bias in the outcome. For example, cDNA fragmentation is usually strongly biased towards the identification of sequences from the 3' ends of transcripts, while RNA fragmentation has little bias over the transcript but is depleted for transcript ends. Therefore, cDNA fragmentation provides valuable information about the precise identity of these ends and RNA fragmentation provides access to precisely identity of the transcript body.
In the classic NGS protocols, adapters are ligated onto shared double-stranded DNA fragments. However, a major drawback of this approach is the loss of information on transcriptional direction. Pre-treat the RNA samples with sodium bisulphate can convert the cytidine into uridine. Widespread C-T transition thereby marks the coding stand of each transcript. Some other methods that maintain strand-specificity have been proposed, such as direct ligation of RNA adaptors to the RNA sample before reverse transcription.
Services you may interested in
The RNA-seq is currently dominated by three different platforms: Illumina (Genome Analyzer and HiSeq), Applied Biosystems SOLID, and Roche 454 Life Science systems. Read lengths range from 30-100 bp for Illumina and SOLiD, and 200-500 bp for 454 pyrosequencing system. 454-based RNA-seq is particularly attractive for non-model organisms without reference genomes or transcriptomes. Longer reads or paired-end short reads can reveal connectivity between multiple exons. RNA-seq is a powerful method to study complex transcriptomes and reveal sequence variations in the transcribed regions.
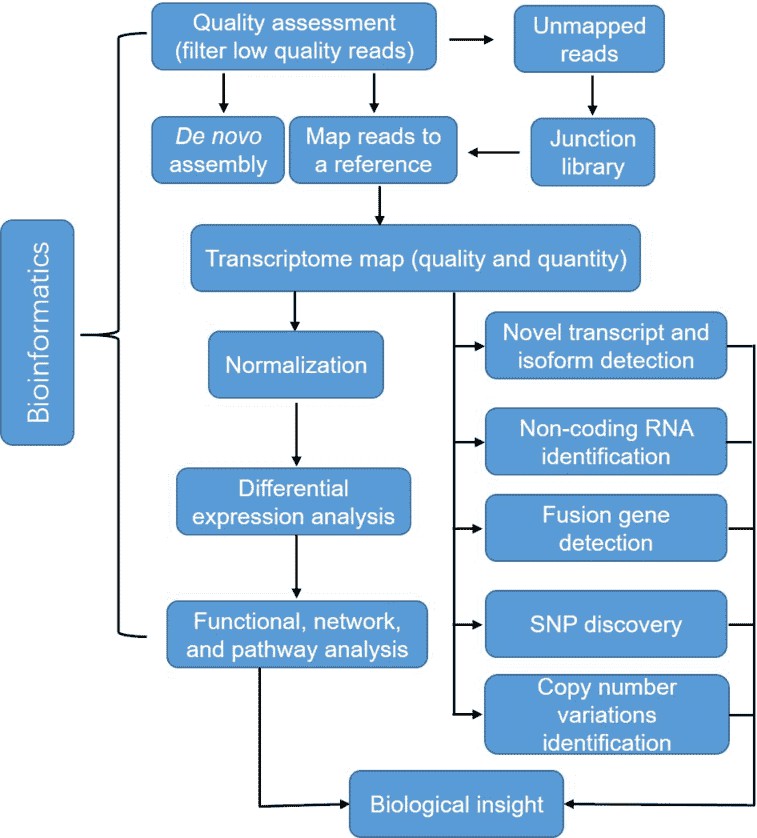 Figure 3. A typical analysis pipeline of RNA-seq data.
Figure 3. A typical analysis pipeline of RNA-seq data.
Quality assessment is the first step for the bioinformatics analysis of RNA-seq, which ensures a coherent final result by removal of low-quality sequences, over-represented sequences, and adapter sequences. Once all reads have been filtered and mapped or assembled, gene expression levels can thus be inferred, leading to a genome-scale transcriptome map in terms of quality and quantity. RNA-seq also allows detecting differential expression (DE) across treatments of conditions. Normalization has to be conducted to adjust the differences between samples such as library size and gene-specific features. Furthermore, RNA-seq enables us to identify SNPs, fusion genes, and post-transcriptional gene regulation, such as RNA editing, degradation, and translation.
If you want more information about the applications of RNA-seq or bioinformatics workflow of RNA-seq, you can refer to the article.
References:
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews genetics, 2009, 10(1): 57.
- Qian X, Ba Y, Zhuang Q, et al. RNA-Seq technology and its application in fish transcriptomics. Omics: a journal of integrative biology, 2014, 18(2): 98-110.
- Marguerat S, Bähler J. RNA-seq: from technology to biology. Cellular and molecular life sciences, 2010, 67(4): 569-579.
- Wilhelm B T, Landry J R. RNA-Seq—quantitative measurement of expression through massively parallel RNA-sequencing. Methods, 2009, 48(3): 249-257.
- McGettigan P A. Transcriptomics in the RNA-seq era. Current opinion in chemical biology, 2013, 17(1): 4-11.

 Sample Submission Guidelines
Sample Submission Guidelines


