
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

Regulation of gene expression is fundamental to link genotypes with phenotypes. RNAs shape complex gene expression networks which drive biological processes. An in-depth understanding of the underlying mechanisms about how to govern these complex gene expression networks is vital for the treatment of complex disease such as cancer. Hybridization-based microarrays are used to allow the simultaneous monitoring of expression levels of annotated genes in cell populations. However, genome-wide approaches are proved to provide more valuable insights into transcriptomes. These next/third sequencing platforms allow the rapid and cost-effective generation of massive amounts of sequence data. The RNA profiling by utilizing high-throughput sequencing technologies are known as RNA-seq.
Services you may interested in
Since RNA-seq is quantitative, it is useful to determine RNA expression levels. In addition to this basic function, RNA-seq can be used for differential gene expression, variants detection and allele-specific expression, small RNA profiling, characterization of alternative splicing patterns, system biology, single-cell RNA-seq and developing SNPs and SSRs etc.
RNA-Seq, leveraging high-throughput sequencing technology, enables comprehensive cDNA library sequencing of all RNAs transcribed within a cellular or tissue sample. This approach allows for the quantitative assessment of specific RNA expression by counting corresponding read numbers, aiding in the discovery of novel transcripts. Provided there is a reference genome, these transcripts can be mapped back to the genome, enhancing the understanding of comprehensive genetic information, such as transcript location and splicing patterns. This widely applied technique is integral to various fields, including biological research, medical studies, clinical research, and pharmaceutical development, significantly enriching our understanding and application of genomic sciences.
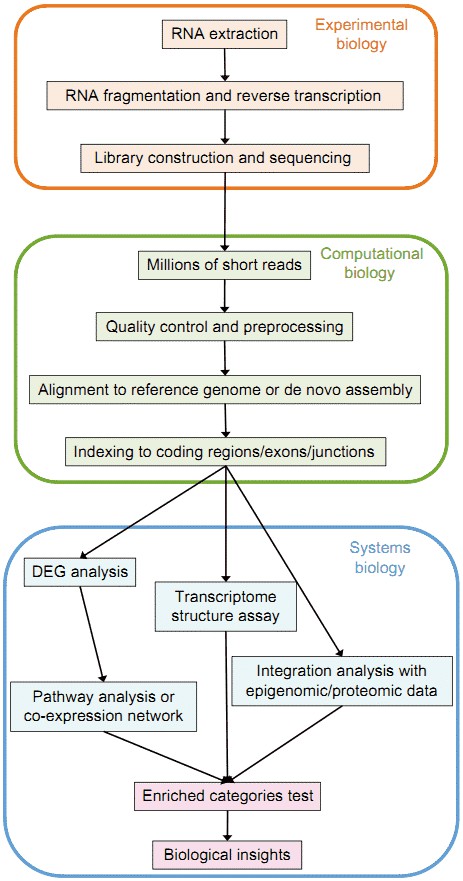 Figure 1. Overview of the typical RNA-seq analysis pipeline (Han et al. 2015).
Figure 1. Overview of the typical RNA-seq analysis pipeline (Han et al. 2015).
Differential gene expression
An important application of RNA-seq is the comparison of transcriptomes across different developmental stages, treatments, or disease conditions. This analysis, also known as differential gene expression analysis, requires identification of genes along with their isoforms and precise assessment of their expression levels. It is important to illustrate functional elements of the genome and uncover the biological mechanisms of development and disease.
The common tools for differential gene expression include Cuffdiff, DESeq, DESeq2, EdgeR, PoissonSeq, Limma voom, and MISO.
Variants detection and allele-specific expression
RNA-seq allows identification of variants and allele-specific expression. Single-nucleotide polymorphisms (SNPs) refer to the variation in a single nucleotide that occurs at a specific position in the genome, which may lead to allele-specific expression (ASE). ASE means that one of two alleles is highly transcribed into mRNA and the other is lowly transcribed or even not transcribed at all. Recent studies have also associated ASE to the susceptibility of a number of human diseases. RNA-seq and whole-genome DNA sequencing (WGS) allow identification of common disease variants, including SNPs and ASE.
The common tools used for variants detection are GATK, ANNOVAR, SNPiR, SNiPlay3.
Small RNA profiling
Small RNA species generally involve microRNA (miRNA), small interfering RNA (siRNA), and piwi-interacting RNA (piRNA), as well as other types of small RNA, such as small nucleolar RNA (snoRNA) and small nuclear RNA (snRNA). Small RNAs play a role in gene silencing and post-transcriptional regulation of gene expression. Small RNAs have been demonstrated to be involved in biological processes, including development, cell proliferation and differentiation, and apoptosis. Most initial small RNA discovery studies used pyrosequencing, and subsequently, other NGS platforms with higher throughput, which resulted in genome-wide surveys and the discovery of an increasing number of small RNA species. Common bioinformatic tools for small RNA sequencing data are shown in Table 1.
Table 1. sRNA-seq web application comparison (Rahman et al. 2018).
| Features | Oasis 2 | omiRas | mirTools 2.0 | MAGI | Chimira | sRNAtoolbox |
| FASTQ compression | √ | √ | √ | |||
| miRNA modifications and edits | √ | √ | √ | √ | √ | |
| Novel miRNA database | √ | √ | ||||
| Infection and cross-species analysis | √ | |||||
| Non-model organism | √ | √ | ||||
| Differential expression | √ | √ | √ | √ | √ | √ |
| Multivariate differential expression | √ | √ | ||||
| Classification | √ | |||||
| Novel miRNA target prediction | √ | √ | √ | |||
| Pathway/GO analysis | √ | √ | √ | √ | √ | |
| Batch job submission (API) | √ | |||||
| Genome browser | √ |
Characterization of alternative splicing patterns
Alternative splicing patterns are important to understand development and human diseases since altered splicing patterns contribute to development, cell differentiation, and human disease. RNA-seq is a powerful tool for characterization of alternative splicing patterns. Paired-end sequencing enables sequence information from both ends, thereby detecting splicing patterns without a requirement for previous knowledge of transcript annotations. PacBio SMRT sequencing allows examination of splicing patterns and transcript connectivity in an unbiased and genome-scale manner by generating full-length transcript sequences.
The common tools for characterization of alternative splicing patterns include TopHat, MapSplice, SpliceMap, SplitSeek, GEM mapper, SpliceR, SplicingCompass, GIMMPS, MATS, and rMATS.
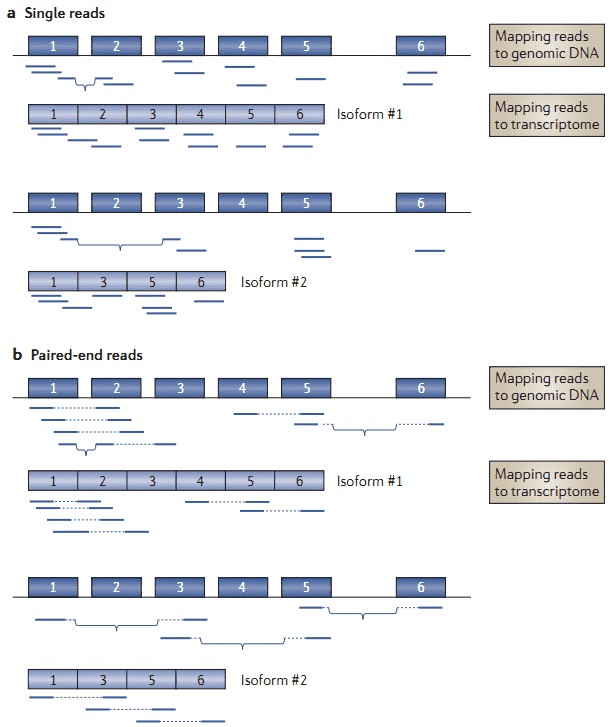 Figure 2. RNA-seq for detection of alternative splicing events (Ozsolak and Milos 2011).
Figure 2. RNA-seq for detection of alternative splicing events (Ozsolak and Milos 2011).
System biology
Creating lists of differential expression (DE) genes is not the final step of RNA-seq analysis. Further biological insight into an experimental system can be acquired by looking at the expression changes of sets of genes. This process, known as system biology, is based on the understanding that the whole is greater than the sum of the parts. Pathway analysis and co-expression network analysis are two important included parts.
Table 2. The tools for pathway analysis and co-expression network analysis using RNA-seq data.
| Pathway analysis | GSEA | A knowledge-based approach for genome-wide expression profiling. |
| GSVA | A non-parametric, unsupervised method for estimating variation of gene set enrichment through the samples of an expression data set. | |
| SeqGSEA | Provides methods for gene set enrichment analysis by integrating differential expression and splicing. | |
| GAGE | An evaluation of the very latest large-scale genome assembly algorithms. | |
| SPIA | Identifies the pathways most relevant to the condition | |
| TAPPA | A java-based tool for identification of phenotype-associated genetic pathways. | |
| DEAP | Identifies important regulatory patterns from differential expression data. | |
| GSAASeqSP | Can identify pathways or gene sets significantly ass | |
| Co-expression network | GSCA | help researchers make discoveries by using massive amounts of publicly available gene expression data. |
| DICER | Detects differentially co-expressed gene sets by using a novel probabilistic score for differential correlation. | |
| WGCNA | A powerful method to isolate co-expressed groups of genes from microarray or RNA-seq data. |
Single-cell RNA-seq
The single-cell RNA-seq offers opportunities to dissect of the interplay between intrinsic cellular processes and extrinsic stimuli in cell fate determination. It also contributes to a better understanding of how an 'outlier cell' may determine the outcome of an infection. In addition, a majority of living cells cannot be cultivated in vitro, single-cell RNA-seq may discover novel species or regulatory processes of biotechnological or medical relevance. The workflow of single-cell RNA-seq generally involves the following steps: single-cell isolation, cDNA library construction, RNA-seq, and bioinformatics (Figure 2).
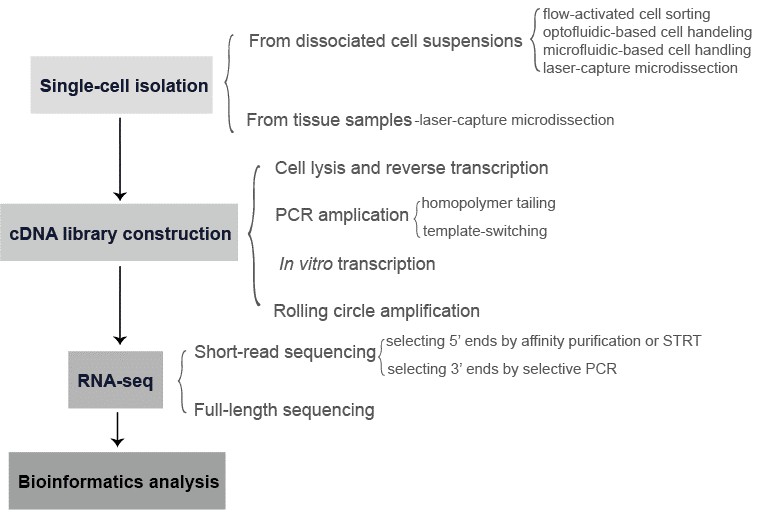 Figure 3. The general workflow of single-cell RNA-seq.
Figure 3. The general workflow of single-cell RNA-seq.
Applications of single-cell RNA-seq:
Developing SNPs and SSRs
RNA-seq is a powerful tool that can be utilised to detect and identify single-nucleotide polymorphisms (SNPs) within genomes. By comparing and analysing the transcriptomic data of different individuals or populations, we can pinpoint SNP locations within various genomes and subsequently screen and validate these. This process aids our comprehension of genetic divergence between individual organisms, providing a solid foundation for further exploration into the relationships between genotypes and phenotypes. Moreover, RNA-seq can also be employed to discover and scrutinise Simple Sequence Repeats (SSRs) within the genomic structure. SSRs, consist of repeated DNA sequences found within the genome, are paramount to genetic marking and population genetics research. By analysing RNA sequence data, we can identify genes and transcripts containing SSRs, thereby laying important groundwork for studies in genetic diversity and evolution.
Utilizing RNA-seq data, we are capable of constructing a genetic map of the genome-- thereby revealing the distribution of gene loci and the extent of genetic diversity. Through the analysis of RNA sequences from distinct individuals or populations, we are not only able to establish a connection between genotype and phenotype, but also infer the function of SNPs and SSRs within the genome, alongside their evolutionary significance. RNA-seq also empowers us to identify and authenticate the genetic differences between diverse germplasm resources and varieties. By comparatively analyzing the transcriptome data from different germplasms or varieties, we can uncover SNPs and SSRs that carry specific genotypic or phenotypic traits. Hence, this lays a foundation for the collection, preservation, and utilization of germplasm resources.
RNA-seq has been employed in various aspects of cancer research and therapy, encompassing the discovery and characterization of biomarkers detailing cancer heterogeneity and evolution, drug resistance, immune microenvironments in cancer and immuno-therapy, and neoantigens. Notably, gene fusions have been inextricably linked with tumorigenesis and serve as ideal cancer biometrics and therapeutic targets, predominantly detected through RNA-CaptureSeq in clinical samples. Besides nucleic acid biomarkers, the integration of RNA-seq with immunohistochemistry and protein blots has confirmed the identification of certain proteins as cancer biomarkers. For instance, the combination of nuclear COX2 (Cyclooxygenase-2) and HER2 (Human Epidermal Growth Factor Receptor 2) has emerged as a potential biomarker for cancer, specifically in diagnosing colorectal cancer and predicting its prognosis. RNA-seq can detect early-stage mutations and high-risk polymorphisms, leading to the uncovering of novel cancer biomarkers and potential therapeutic targets, thereby monitoring diseases and guiding targeted therapy in early treatment decisions. For instance, in breast cancer, single-cell RNA-seq unveiled that tumor-infiltrating immunosuppressive immature myeloid cells contribute to drug resistance.
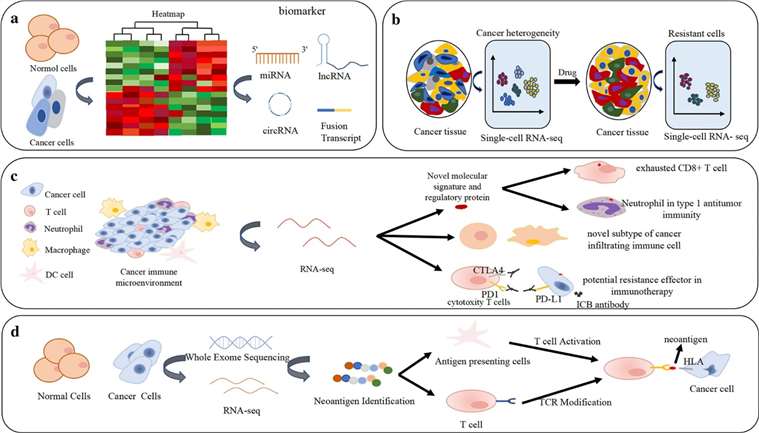 Figure 4. Applications of RNA-seq in differential expression analysis and cancer biomarkers, cancer heterogeneity and drug resistance, cancer immune microenvironment, immunotherapy and neoantigen. (Hong et al., 2020)
Figure 4. Applications of RNA-seq in differential expression analysis and cancer biomarkers, cancer heterogeneity and drug resistance, cancer immune microenvironment, immunotherapy and neoantigen. (Hong et al., 2020)
Through the application of RNA-sequencing (RNA-seq), we can contribute to the unveiling of mechanisms underlying disease onset, enhance precision in disease diagnostics, evaluate treatment efficacy, and identify novel treatment targets. With a comparative analysis of transcriptomic data from patient samples and standard control groups using RNA-seq, it's possible to discern differences in gene expression associated with disease onset and progression, thereby revealing potential disease mechanisms. In the identification of biomarkers related to various diseases, the role of RNA-seq is vital. By examining the transcriptional data of patient samples, specific patterns of gene expression correlated with disease states can be uncovered. This revelation of novel biomarkers has substantial implications for early disease diagnostics and prognostic assessment. Furthermore, RNA-seq can be employed to pinpoint new therapeutic targets, guide personalized treatment plans, and inform immunotherapeutic strategies. Apart from its usage in cancer research, RNA-seq has found wide applications in the study of numerous other diseases, including cardiovascular afflictions, neurological disorders, and autoimmune diseases. The examination and analysis of patient-derived transcriptomic data pave the way to a deeper understanding of disease mechanisms which in turn provide fresh perspectives and strategies for disease diagnosis and treatment.
Employing RNA-Seq, a high-throughput sequencing technology, carries substantial implications across various biological fields. Applications of RNA-seq in biology mainly encompass gene expression analysis, non-coding RNA research, epigenetic studies, molecular evolution research, and functional genomics research. By measuring mRNA expression levels at the transcriptome level, differentially expressed genes under varying conditions can be identified, unveiling gene regulatory networks and signal pathways. RNA-seq can facilitate the discovery and analysis of non-coding RNAs, such as miRNAs and lncRNAs, which play essential roles in gene regulation, epigenetics, and disease occurrence. Regarding molecular evolution research, RNA-Seq data can be used to compare gene expression spectrums amongst different species or populations, thereby investigating gene function evolution and differentiation. Moreover, RNA-seq serves to identify and functionally interpret functional genes and regulatory elements within the genome.
Utilizing RNA-seq data, in-depth research into microbial pathogenic mechanisms can be undertaken. RNA-seq allows us to construct transcriptional regulatory networks in microbes and uncover the relationships between regulatory factors and pathogenicity-associated genes. The identification of genes encoding virulence factors and immune evasion mechanisms in microbes significantly aids our understanding of pathogenesis, as these genes often play crucial roles in microbial invasions and immune system circumvention. Importantly, RNA-seq can also facilitate research into microbial resistance to antibiotics. An analysis of microbial transcriptomic changes following antibiotic treatment can shed light on the genes and pathways involved in resistance development. Moreover, this technology can examine the gene expression alterations in host cells during a microbial infection process, which can reveal insights into microbial invasion mechanisms, host immune responses, and molecular interactions involved in the pathogenic process.
RNA-Seq has found profound applications in agriculture, particularly in studying animal and plant diseases, investigating crop resistance and adaptability, as well as advancing molecular breeding. Through RNA-Seq, we can assess gene expression changes in infected animal and plant tissues to identify disease resistance-related genes. Additionally, RNA-Seq enables the exploration of physiological and metabolic shifts during the onset of disease, elucidating its pathophysiology. RNA-Seq can be instrumental when unraveling the molecular mechanisms through which crops counteract environmental stresses. By investigating gene expression changes in crops under adversarial conditions, we can identify stress-resistance associated genes. This adds theoretical backing to the selective breeding of new crop variations that are adapted to diverse environmental conditions. Furthermore, RNA-Seq allows for the analysis of the correlation between extensive gene expression data and phenotypic trait data, thereby facilitating genotype-phenotype association studies. This is pivotal in identifying functional genes associated with target traits, providing potential molecular markers and genetic background information for molecular breeding.
The results of RNA-Seq analysis can be leveraged to develop molecular markers for use in marker-assisted selection (MAS) in crop breeding. This approach aids in enhancing breeding efficiency and expediting the selection and dissemination of favorable genes. Additionally, RNA-seq can elucidate changes in gene expression during the interaction between plants and pathogenic microorganisms or pests, contributing to the understanding of the molecular mechanisms underlying plant disease and pest resistance. This provides novel targets and strategies for disease and pest management. RNA-seq is also employed to investigate the transcriptional regulatory networks involved in the interaction between plants and beneficial microorganisms. It helps identify the promoting effects of biofertilizers on plant growth and nutrient uptake, thereby enhancing crop yield and quality.
If you want more information about RNA-seq, please refer to the following articles:
Bioinformatics workflow of RNA-seq
The technologies and workflow of RNA-seq
References:


