Epigenomics: Overview and Technological Advancements
Epigenomics represents a comprehensive approach to studying epigenetic phenomena across the entire genome. This field extends beyond traditional epigenetics by examining regulatory mechanisms at a global scale, leveraging high-throughput technologies to generate genome-wide profiles of various epigenetic marks.
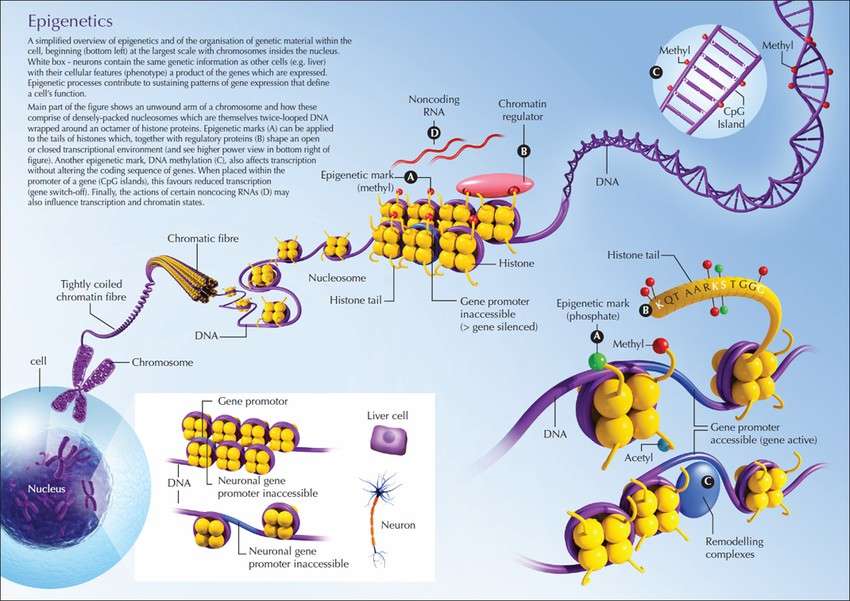 Fig. 1 Overview of epigenetic mechanisms. (Katja Kobow et L,.2020)
Fig. 1 Overview of epigenetic mechanisms. (Katja Kobow et L,.2020)
Key areas of focus in epigenomics include:
Genome-wide DNA methylation patterns
Global histone modification landscapes
Chromatin accessibility profiles
Non-coding RNA distributions
RNA modification levels
Multi-dimensional regulatory mechanisms at the translational level
Recent advancements in epigenomics have been driven by four main areas:
Technological innovations
Exploration of novel epigenetic concepts
New biological applications
Investigation of established molecular mechanisms and pathways
Emerging Technologies and Concepts
Several cutting-edge technologies and concepts are shaping the field of epigenomics:
HiChIP: A method combining chromatin immunoprecipitation with proximity ligation to map protein-centric chromatin interactions
CUT&Tag: A technique for efficient profiling of chromatin proteins, histone modifications, and nucleosomes in low cell numbers
ARTR-seq/LACE-seq: Novel approaches for mapping RNA-chromatin interactions
Active Ribo-seq: A method for profiling actively translating ribosomes
Single-Cell Hi-C: Technique for analyzing 3D genome organization at the single-cell level
Novel Epigenetic Concepts
Researchers are exploring new epigenetic phenomena, including:
Histone lactylation
Super-enhancers
G-quadruplex structures
R-loops
Novel DNA/RNA/protein modifications
Biological Applications and Mechanistic Studies
Epigenomics is providing insights into various biological processes and disease mechanisms:
DNA repair and chemotherapy resistance in relation to histone lactylation
Memory T cell formation involving novel histone demethylases
Transgenerational inheritance mediated by DNA methylation
Gene therapy approaches targeting R-loops
Mechanistic studies are uncovering complex regulatory pathways, such as:
Histone lactylation's influence on the Warburg effect via KRAS
Super-enhancer regulation of METTL3 and its impact on EGFR m6A modification
FTO-mediated m6A modification of HIF1A and its effects on translation
These advancements in epigenomics are providing a more nuanced understanding of gene regulation and cellular function, with potential implications for disease treatment and personalized medicine.
Approaches to Defining Epigenetic Research Questions
To address specific epigenetic questions with greater precision, two distinct approaches can be taken depending on the focus of the research:
Application-oriented Research: When the research emphasizes distinct biological manifestations or case studies, the investigation often begins with a notable phenotype. During the course of the study, sufficient evidence is gathered to elucidate the underlying biological mechanisms behind the observed phenotype. Such research typically forms the basis for grant applications or the publication of novel findings.
Basic Research and Molecular Mechanism Studies: For studies focused on the fundamental molecular mechanisms, particular attention is paid to the role of specific genes within pathways or processes. Research in this domain requires clear identification of downstream targets and focuses on how epigenetic factors regulate these genes or vice versa. The ultimate goal is to understand how these mechanisms influence downstream biological phenomena.
Integrating Epigenomics with Other Omics Approaches
In addition to focusing on specific biological or molecular mechanisms, epigenomics is increasingly being integrated with other omics approaches, such as genomics, transcriptomics, and proteomics. This integrated approach allows for a more comprehensive understanding of the complex interactions between various layers of biological regulation. The following sections will discuss the strategies for combining epigenomics with other omics technologies, providing a deeper insight into the broader biological systems in which these epigenetic modifications occur.
Epigenetic Research: Integration with Other Omics Technologies
When the specific epigenetic domain to be studied has been identified, such as histone acetylation, the inhibitory effects of transcription factors, or RNA m6A modification, these typically become the core focus of the research. Other omics technologies often serve as auxiliary tools, facilitating the analysis of how these epigenetic modifications impact downstream genes or pathways. In some cases, prior omics analyses may uncover key epigenetic factors, thereby providing direction for subsequent research.
In instances where the specific epigenetic focus remains undefined, screening-based omics technologies become critical. For example, transcriptome sequencing and metabolomics can identify abnormalities in epigenetic regulators or associated metabolites. Transcriptome sequencing can analyze gene expression related to transcription factors, histone modifications, RNA modifications, DNA modifications, and protein modifications, including the RNA levels of modifying or demodifying enzymes. Metabolomics, on the other hand, can detect metabolites such as acetyl-CoA and lactyl-CoA, which may suggest the need for further investigation into protein or histone modifications.
Once the epigenetic focus has been determined, the research typically divides into two primary categories: (1) the elucidation of specific molecular mechanisms and (2) the investigation of downstream biological phenomena or functions.
In recent years, epigenomics has increasingly integrated with other omics fields, leading to the emergence of several key research directions: the combination of epigenomics with other epigenomic approaches, the integration of epigenomics with conventional bulk omics, and the current trend of combining epigenomics with single-cell omics. These integrative approaches provide new possibilities and tools for advancing epigenomic research.
Epigenomics Integration: Analytical Strategies and Case Studies
Common Combinations in Epigenomics Research
The integration of multiple epigenomic methodologies has become increasingly prevalent, with several commonly employed combinations, including:
Analytical Strategies in Multi-Omics Approaches
Two principal strategies are commonly employed in multi-omics data integration to investigate epigenomic mechanisms:
1. Direct Correlation Analysis
This approach involves identifying potential research targets by analyzing correlations between datasets from two or more omics platforms. For example:
- Intersecting Candidate Genes: Potential target genes can be screened within a single omics dataset and further refined by intersecting results with another dataset.
Direct correlation analysis is particularly useful for rapidly narrowing the scope of investigation to focus on promising research candidates.
2. Indirect Validation Method
This strategy examines the regulatory hierarchy, focusing on upstream and downstream interactions. For instance:
- Investigating transcription factors or histone modifications that initiate downstream processes such as gene transcription, post-transcriptional modifications, and translation.
- By validating upstream-downstream relationships, this method allows for a more nuanced understanding of regulatory mechanisms.
The direct correlation and indirect validation approaches provide complementary perspectives and methodologies. They cater to distinct research objectives and experimental needs, enabling tailored investigations into epigenomic regulation. The integration of these strategies enriches the capacity for uncovering complex regulatory networks, thereby advancing the understanding of epigenetic contributions to biological processes.
Case Studies in Multi-Omics Approaches for Epigenomic Research
Case 1: Translationomics and m6A Sequencing Reveal YTHDF1-Mediated Regulation of Autophagy-Related Genes in Hepatocellular Carcinoma
Title: Hypoxia-Induced Expression of m6A Reader YTHDF1 Drives Autophagy and Malignancy in Hepatocellular Carcinoma by Promoting ATG2A and ATG14 Translation
A combined analysis of m6A sequencing and proteomics identified only two autophagy-related genes, ATG2A and ATG14, as critical targets. Further investigation using polysome profiling demonstrated that YTHDF1 facilitates the transfer of ATG2A and ATG14 mRNAs to polysomes, thereby increasing their translation efficiency and elevating corresponding protein levels. While translationomics served as a validation tool in this study, m6A sequencing was the central technique, emphasizing its pivotal role in elucidating the regulatory mechanisms of autophagy under hypoxic conditions.
Case 2: Multi-Omics Integration of m6A Sequencing, Translationomics, and RIP-seq Reveals Mechanisms of Meiotic Progression During Spermatogenesis
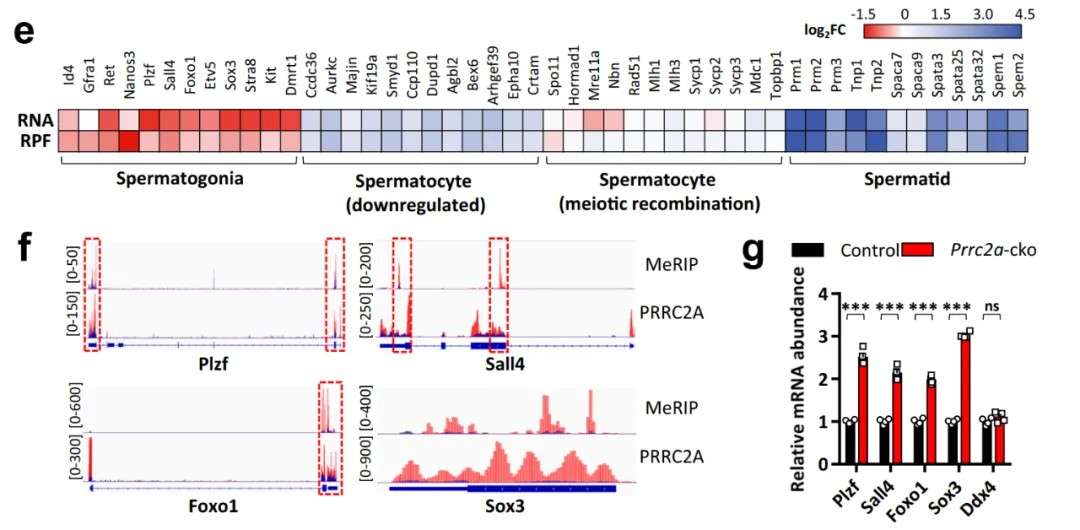
Title: The m6A Reader PRRC2A is Essential for Meiotic Completion During Spermatogenesis
A comprehensive analysis integrating transcriptomics and translationomics identified four functional modules of upregulated and downregulated genes in spermatogonia. Subsequently, a combination of RIP-seq and m6A sequencing revealed 3,366 shared peaks, which were further intersected with transcriptomic and translationomic data. Key upregulated genes, including Plzf, Sall4, and Foxo1, critical for spermatogonial stem cell maintenance, were identified alongside downregulated genes such as Sox3, essential for spermatocyte differentiation. This integrative approach demonstrated the mechanistic links between m6A modifications and the regulation of meiosis.
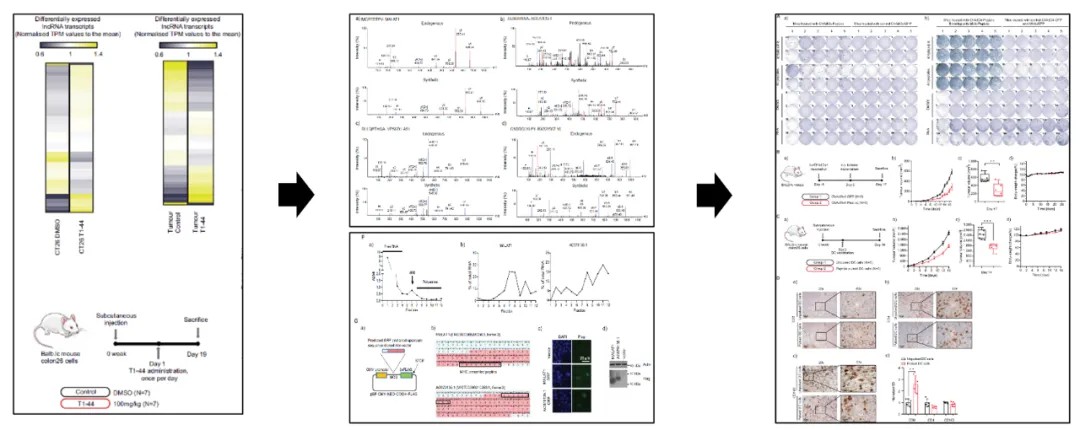
Case 3: lncRNA Sequencing, Translationomics, and Proteomics Reveal Immunogenic Peptides Encoded by lncRNAs Driving Anti-Tumor Responses
Title: Long Non-Coding RNA-Derived Peptides are Immunogenic and Drive Potent Anti-Tumor Responses
Treatment of murine colorectal cancer cells with therapeutic agents resulted in suppressed tumor growth and a widespread downregulation of lncRNAs. Mass spectrometry and polysome profiling analyses demonstrated that certain lncRNAs encode immunogenic peptides that bind MHC class I molecules, activating anti-tumor immune responses. The encoded peptides significantly inhibited tumor growth and exhibited potent immunogenicity, capable of stimulating the host immune system. Additionally, a viral vector-based lncRNA vaccine strategy was shown to delay tumor progression, highlighting the therapeutic potential of lncRNA-derived peptides in cancer immunotherapy.
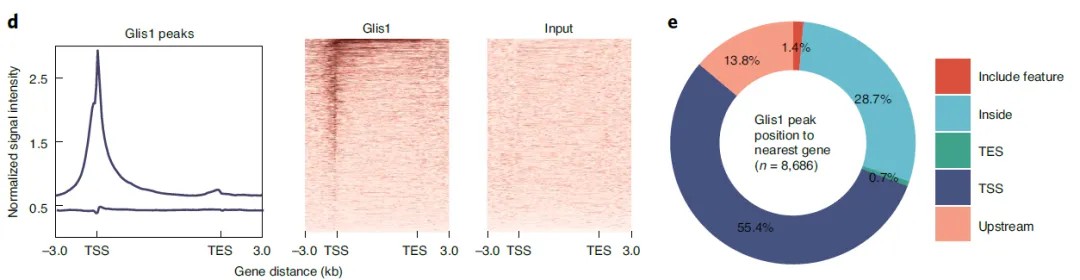
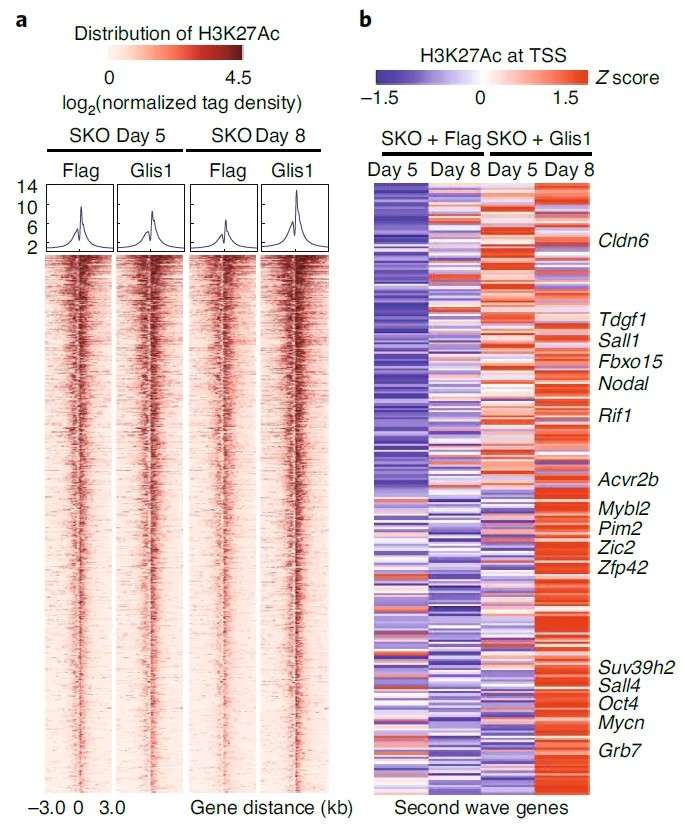
Case 4: Combined ChIP-seq and ATAC-seq Analysis Reveals Glis1 Activation of Epigenomic Mechanisms During Cell Reprogramming
Title: Glis1 Facilitates Induction of Pluripotency via an Epigenome-Metabolome-Epigenome Signaling Cascade
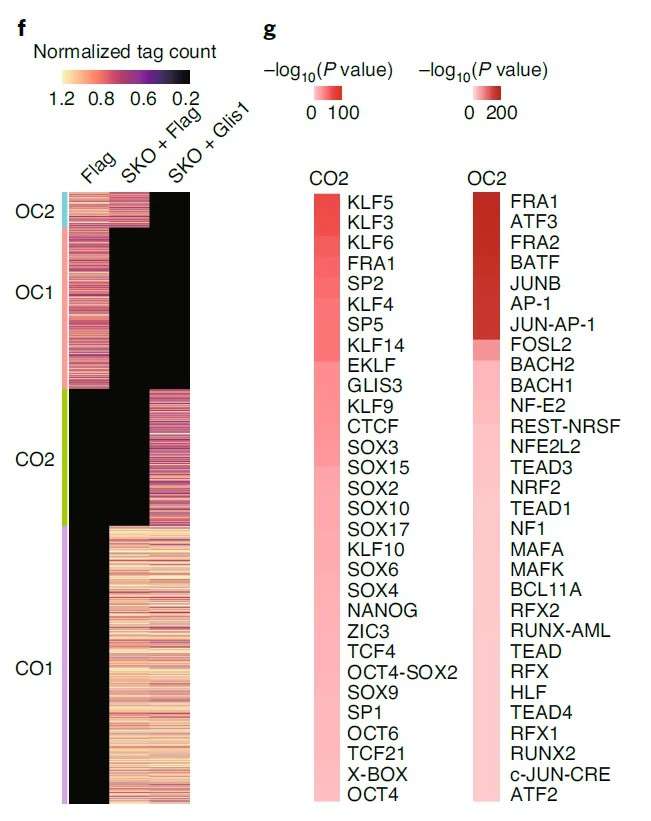
This study employed a multi-omics approach, including ChIP-seq, ATAC-seq, transcriptomics, and metabolomics, to investigate the role of Glis1 in cell reprogramming. ChIP-seq analysis revealed that Glis1 regulates genes involved in glycolysis, a process associated with histone lactylation and activation of the transcriptional coactivator P300. The activation of glycolytic genes was accompanied by significant transcriptional activity. ATAC-seq analysis further identified motifs for key transcription factors, such as KLF and SOX, within chromatin regions opened by Glis1. Overexpression of Glis1 promoted chromatin accessibility at pluripotency-associated genes and suppressed somatic gene expression, highlighting its dual role in chromatin remodeling and transcriptional activation during reprogramming.
These cases exemplify the utility of integrating multiple omics methodologies to dissect complex regulatory networks in epigenomics, offering insights into mechanisms underlying disease progression, cellular differentiation, and therapeutic responses.
Integration of Epigenomics and Bulk Omics: Analytical Approaches and Case Studies
The combination of epigenomic analyses with conventional bulk omics, including transcriptomics, metabolomics, and proteomics, has proven to be an effective strategy for elucidating complex biological mechanisms. This integration is typically employed in two major scenarios:
1. Preliminary Exploration
In this context, conventional bulk omics serves as an exploratory tool to guide subsequent epigenomic investigations. It facilitates the identification of potential regulatory factors or pathways that warrant further analysis.
2. Validation of Research Findings
Once epigenomic data has been thoroughly analyzed, specific regulatory factors are selected for functional validation through bulk omics techniques. These validation studies are critical for confirming the biological relevance of identified mechanisms.
Role of Specific Techniques in Combined Analyses
ATAC-seq:
- This epigenomic method is particularly suited for large-scale screening of transcription factors.
- Functionally similar to transcriptomics, it is often employed during the initial stages of exploration.
ChIP-seq and CUT&Tag:
- These targeted approaches focus on specific histone modifications or transcription factors, enabling detailed mechanistic studies.
m6A-seq:
- This method specializes in analyzing RNA modifications, specifically N6-methyladenosine (m6A), which is pivotal for post-transcriptional regulation.
Translationomics:
- This technique provides insights into RNA translation activity, allowing the assessment of the functional impact of regulatory elements on protein synthesis.
Advantages of Multi-Omics Integration
The flexible combination of broad-spectrum screening and targeted validation enables researchers to address diverse experimental objectives. By integrating multi-omics approaches, both global trends and specific molecular mechanisms can be systematically examined. This adaptability makes the approach suitable for different stages of research, ranging from hypothesis generation to detailed functional characterization.
 Fig. 2 Transcriptome sequencing + ATAC-seq research strategy
Fig. 2 Transcriptome sequencing + ATAC-seq research strategy
The following sections will present detailed case studies for further clarification of the methods outlined above.
Case Study 1: Transcriptomics, Metabolomics, and ChIP-seq/ATAC-seq Reveal Histone Lactylation and Acetylation in Cellular Reprogramming
This case focuses on the roles of transcriptomics and metabolomics in the epigenetic regulation of cellular reprogramming. The transcriptional profiling of reprogramming cells revealed the upregulation of glycolysis-related genes, including Hk2, Pgk1, Pfkl, Pkm, Eno1, and Ldha, upon Glis1 expression. Conversely, somatic-state marker genes such as Setbp1, Thy1, Il1rn, Prss23, Col6a2, Col6a3, and Col1a1 were markedly downregulated.
Metabolomic analyses conducted on days 5 and 8 of reprogramming using mass spectrometry demonstrated a significant elevation in metabolites associated with the glycolytic pathway (e.g., pyruvate and lactate) and the tricarboxylic acid (TCA) cycle (e.g., acetyl-CoA, citrate, and isocitrate) in the presence of Glis1, particularly on day 8.
Glis1-ChIP-seq combined with transcriptomic data identified upregulated glycolytic genes as being directly regulated by Glis1. Subsequent metabolic profiling highlighted increased substrate levels for lactylation and acetylation, which triggered histone lactylation and acetylation events, as evidenced by ChIP-seq analysis. These modifications affected super-enhancer regions and activated transcription of pluripotency-related genes, exemplifying a hierarchical regulatory cascade from metabolic pathways to chromatin remodeling.
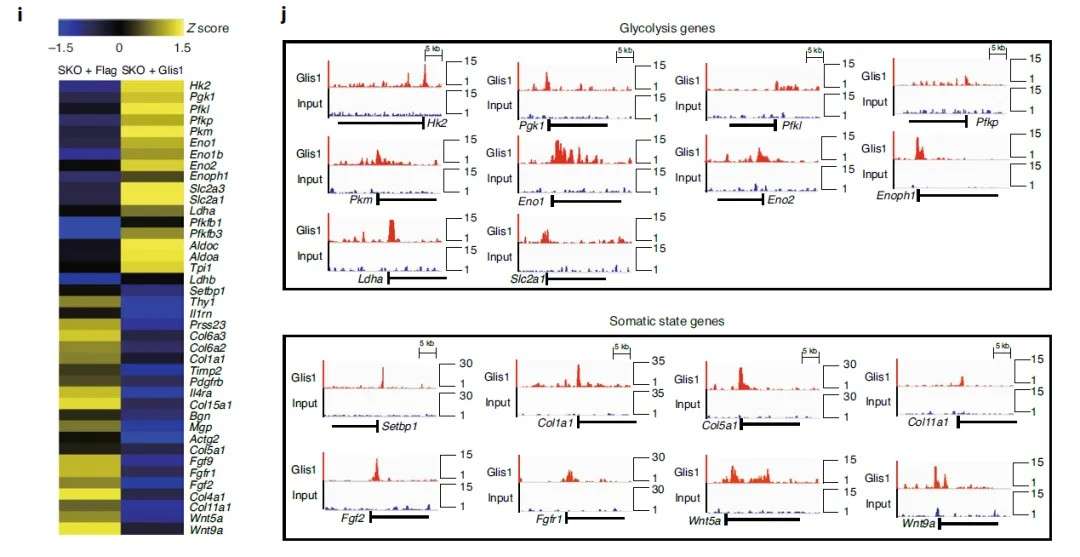
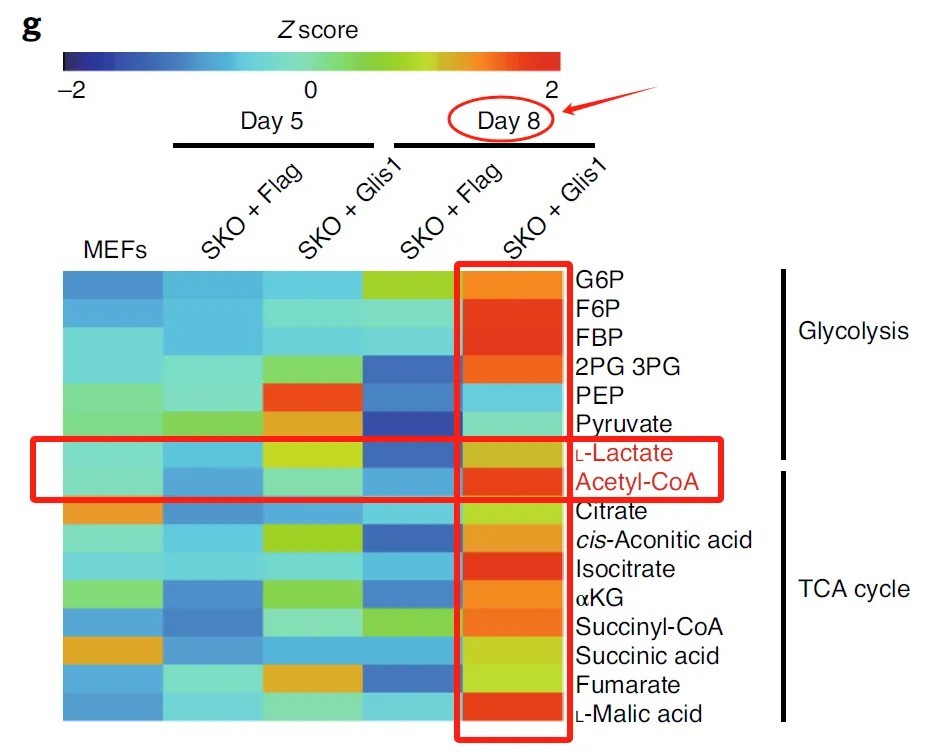
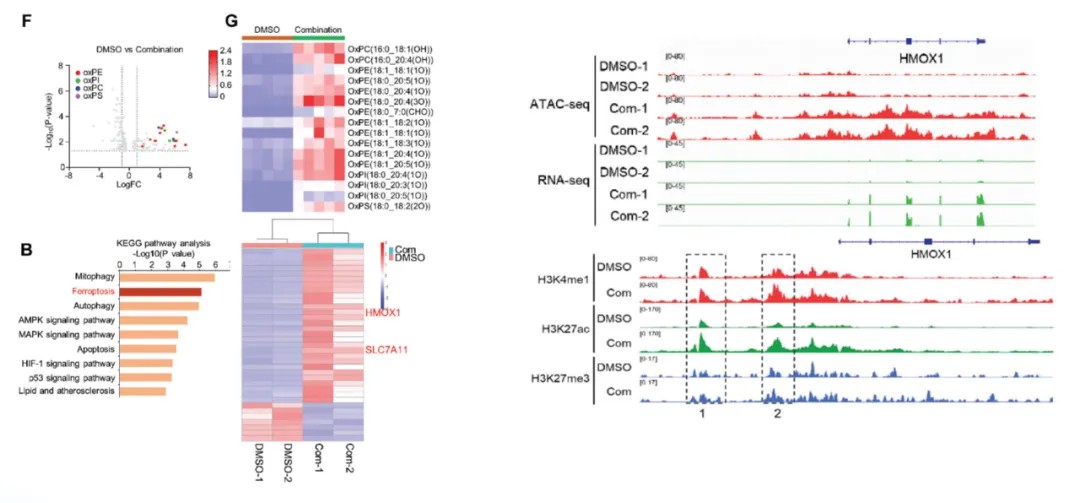
Case Study 2: Metabolomics, Transcriptomics, ATAC-seq, and CUT&Tag Reveal Ferroptosis as a Mechanism of Drug Resistance
Title: Donafenib and GSK-J4 Synergistically Induce Ferroptosis in Liver Cancer by Upregulating HMOX1 Expression
This study employed a multi-omics approach to uncover mechanisms of ferroptosis-mediated drug resistance in liver cancer.
- Metabolomics: The metabolomic analysis pinpointed drug-induced alterations in ferroptosis-associated metabolites, such as lipid peroxides and iron-related compounds.
- Transcriptomics: Differential gene expression analysis identified HMOX1 as a key upregulated gene. The integration of transcriptomics with ATAC-seq using the Integrative Genomics Viewer (IGV) revealed accessible chromatin regions associated with HMOX1.
- Epigenomic Profiling: CUT&Tag analyses were conducted to evaluate the impact of donafenib and GSK-J4 combination therapy on histone modifications in Huh7 cells. Significant changes were observed in histone marks, including H3K27me3, H3K27ac, and H3K4me1, at both enhancer regions and the HMOX1 locus.
These findings demonstrated that ferroptosis induction is mediated by epigenomic modifications, which activate the transcription of ferroptosis-associated genes. Furthermore, the study underscored the synergistic effects of donafenib and GSK-J4, offering insights into therapeutic strategies to overcome drug resistance in hepatocellular carcinoma.
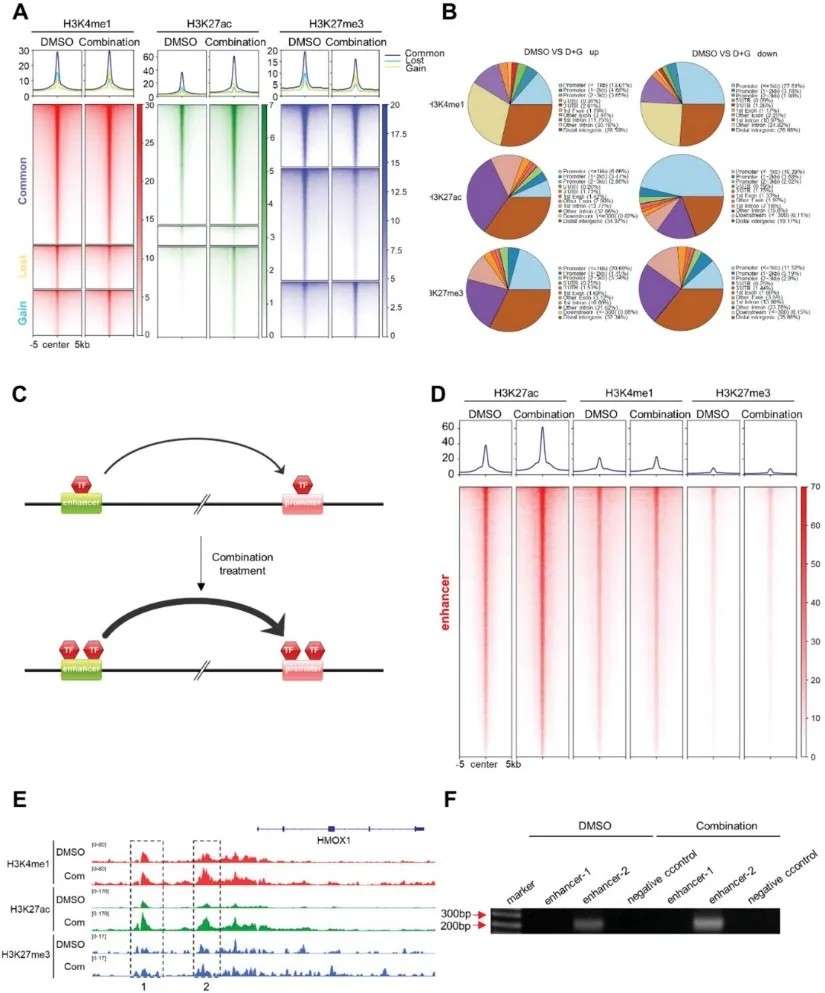
These case studies exemplify the power of multi-omics integration in delineating complex regulatory mechanisms. By combining transcriptomics, metabolomics, and advanced epigenomic techniques such as CUT&Tag and ATAC-seq, researchers can uncover hierarchical relationships between metabolic pathways, chromatin states, and gene expression. This approach not only enhances the understanding of cellular reprogramming and drug resistance but also provides a framework for the development of targeted therapeutic strategies.
Single-Cell Sequencing and Epigenomics: Analytical Strategies and Case Studies
Single-cell sequencing technology has experienced rapid advancements in recent years, significantly contributing to the progress of numerous research endeavors. Initially, the primary benefit derived from single-cell sequencing was the development of the first wave of technological breakthroughs, leading to the emergence of extensive single-cell atlases. These atlases encompass a wide range of areas, including pan-cancer immune cell maps, developmental cell maps, and comprehensive cell atlases of various diseases.
With continuous technological refinements, single-cell sequencing research has evolved from the early focus on large-scale atlases—aimed at identifying all cellular subpopulations—toward more refined, focused research on smaller, specific subpopulations. For example, the study of subtypes such as Cancer-Associated Fibroblasts (CAFs), Tumor-Associated Macrophages (TAMs), NK T cells, and endothelial cells has become a rapidly growing area of interest.
Currently, the emphasis of single-cell sequencing research has shifted to constructing unique "biological narratives" around specific cell subpopulations, such as elucidating their classical signaling pathways or metabolic regulatory mechanisms. At this stage, the role of epigenomics has become increasingly critical.
Integration of Single-Cell Sequencing and Epigenomics
The typical workflow for integrating single-cell sequencing with epigenomics generally involves the following steps:
Identification of Specific Cell Subpopulations:
Single-cell sequencing is employed to define the target cell subpopulations for the study.
Exploration of Epigenetic Factors:
Epigenetic factors specifically expressed within the identified subpopulations are pinpointed.
Differential Analysis (Optional):
A comparative analysis is conducted between the target subpopulation and other cell subpopulations to identify variations in the expression of epigenetic factors.
Enrichment of Targeted Cells:
Techniques such as sorting or enrichment are utilized to isolate the specific subpopulation of cells, allowing for in-depth epigenomic analysis.
The Role of Epigenomics in Single-Cell Research
Epigenomics can be viewed as the "upstream regulator" of gene expression, akin to the source of a river that governs downstream processes. Epigenetic factors are typically limited in number yet are crucial regulators of cellular functions. Key epigenetic factors include:
- Transcription Factors: Comprising only a few hundred factors, these proteins are integral in regulating gene expression.
- Histone Modifiers and Demodifiers: Involving dozens of enzymes, these play central roles in chromatin remodeling.
- RNA Modifiers (e.g., m6A Regulators): A handful of enzymes modulate RNA modifications, with m6A being a prominent example.
- DNA Methyltransferases: A limited number of core enzymes, usually less than twenty, regulate DNA methylation.
The limited number of these epigenetic factors makes them pivotal in cellular regulation, and they frequently serve as the driving force behind high-impact research publications. As such, they form a core framework for developing effective research strategies that can facilitate the publication of high-quality, high-level papers.
 Figure 3: Conventional Epigenomics Research Strategy
Figure 3: Conventional Epigenomics Research Strategy
When combined with single-cell sequencing, research strategies becomes:
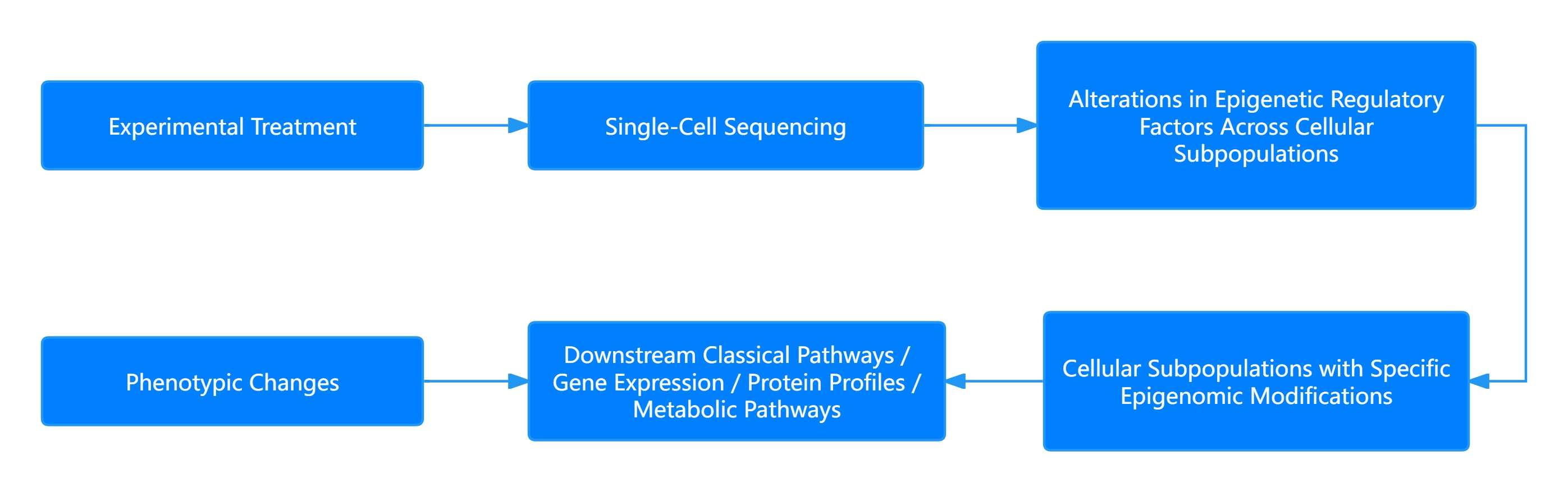 Figure 4: Research Model of Single-Cell Epigenomic Sequencing
Figure 4: Research Model of Single-Cell Epigenomic Sequencing
This figure depicts the research model for single-cell epigenomic sequencing, which integrates single-cell sequencing with epigenomics to analyze epigenetic changes at the cellular level.
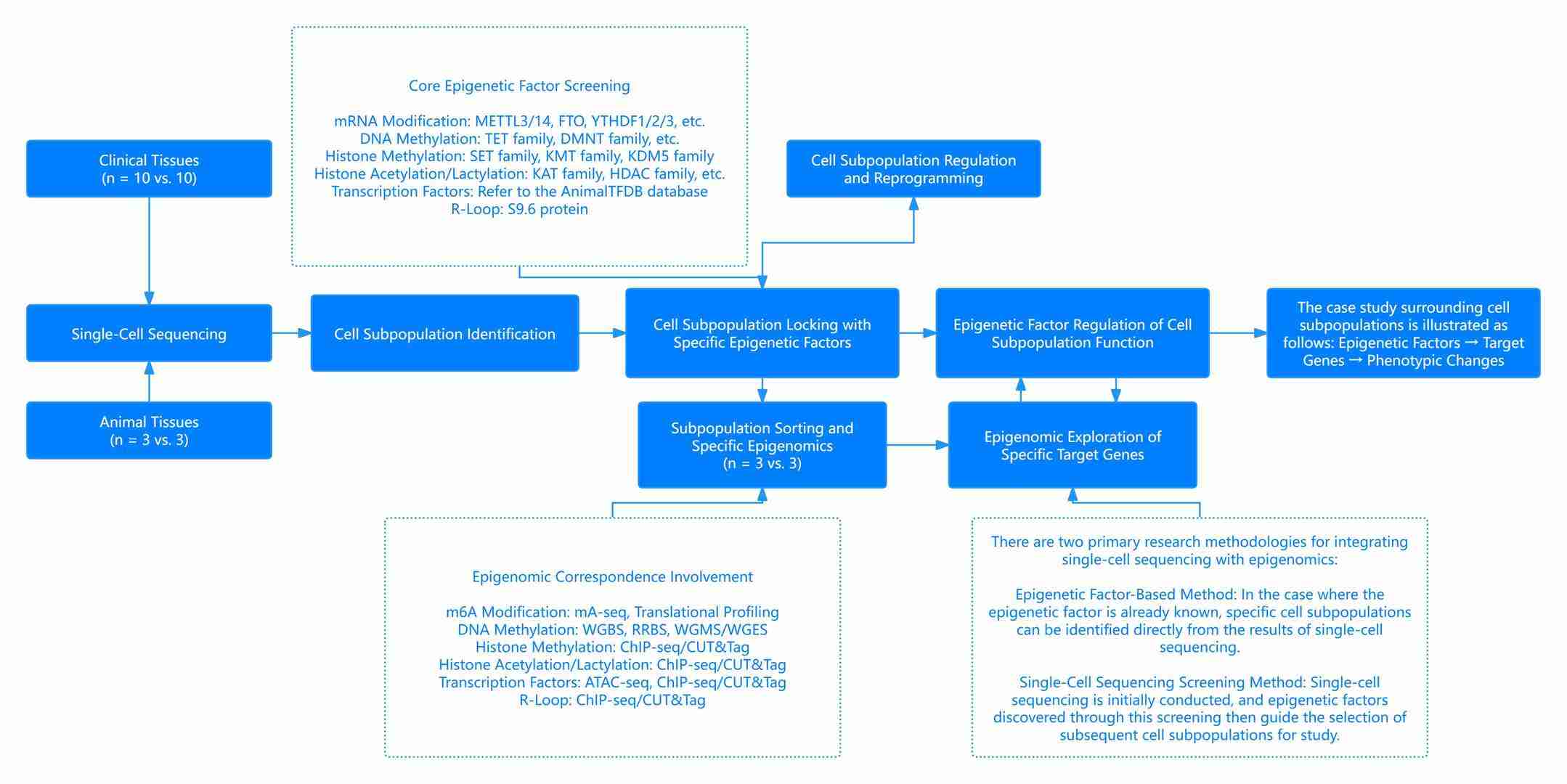 Figure 5: Overall Framework and Conceptual Design of Epigenomic Research Based on Single-Cell Sequencing
Figure 5: Overall Framework and Conceptual Design of Epigenomic Research Based on Single-Cell Sequencing
This figure provides the comprehensive framework and research design for epigenomics based on single-cell sequencing, highlighting the innovative approach and conceptual framework of combining these two fields for deeper insights.
The combination of single-cell sequencing and epigenomics has emerged as a "golden partnership" in high-impact scientific research and represents a novel avenue with substantial innovative potential. The framework and research approach based on single-cell sequencing can be clearly observed in the diagram above.
References:
- Zhang, C., Chen, Y., Sun, B., Wang, L., Yang, Y., Ma, D., Lv, J., Heng, J., Ding, Y., Xue, Y., Lu, X., Xiao, W., Yang, Y. G., & Liu, F. (2017). m6A modulates haematopoietic stem and progenitor cell specification. Nature, 549(7671), 273-276. https://doi.org/10.1038/nature23883
-
Liu, J., Dou, X., Chen, C., Chen, C., Liu, C., Xu, M., Zhao, S., Shen, B., Gao, Y., Han, D., & He, C. (2020). N6-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science, 367(6477), 580-586. https://doi.org/10.1126/science.aay6018
-
Hou, Y., Zhang, H., Miranda, L., & Lin, S. (2010). Serious overestimation in quantitative PCR by circular (supercoiled) plasmid standard: microalgal pcna as the model gene. PloS One, 5(3), e9545. https://doi.org/10.1371/journal.pone.0009545
-
Zaccara, S., Ries, R. J., & Jaffrey, S. R. (2019). Reading, writing and erasing mRNA methylation. Nature Reviews Molecular Cell Biology, 20(10), 608-624. https://doi.org/10.1038/s41580-019-0168-5

 Sample Submission Guidelines
Sample Submission Guidelines


