
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

The Assay for Transposase-Accessible Chromatin using high-throughput sequencing (ATAC-seq) is a groundbreaking technique designed to investigate chromatin accessibility and the underlying mechanisms of gene regulation. A significant advantage of atac sequencing over traditional genomic methodologies is its minimal sample requirement; it can generate high-quality chromatin accessibility data from a remarkably small number of cells, ranging from as few as 500 to 50,000. This makes ATAC-seq a valuable tool for studying small cell populations, different cell types, and genomic variations in limited samples.
At the core of the ATAC-seq technology lies the use of Tn5 transposase, which inserts known adaptor sequences into open regions of chromatin. This process allows for the high-throughput sequencing and subsequent characterization of these accessible regions. Chromatin accessibility is closely related to transcriptional activity. ATAC-seq can map the chromatin structure and identify epigenetic changes in different cell types and under various conditions. Since its introduction in 2013, ATAC-seq has become widely adopted in the fields of gene regulation, epigenomics, and disease research, emerging as a critical instrument for elucidating the molecular basis of complex diseases.
ATAC-seq is useful for both theoretical and practical reasons. In eukaryotes, DNA is highly compacted, unlike in prokaryotes. This compaction starts when DNA wraps around histone proteins to form nucleosomes, which are the basic units of chromatin structure. Subsequent levels of folding give rise to chromatin and chromosomes.
Chromosomes and chromatin represent two states of the same molecular substance. Chromatin exists in a more relaxed, accessible form, whereas chromosomes are tightly coiled. This dynamic transition between states is crucial in regulating gene expression. To enable processes such as DNA replication and transcription, these tightly packed DNA structures must unwind to allow regulatory elements (such as transcription factors or other modulators) to bind effectively. Transcription factors, for instance, identify and attach to specific DNA sequences to initiate the transcription of particular genes. This transformation into a more relaxed chromatin state is referred to as "open chromatin," which is a hallmark of chromatin accessibility. ATAC-seq serves as a pivotal tool in probing these dynamic processes, proving invaluable to scientific research.
Advantages of ATAC-seq Technology
CD Genomics offers a comprehensive range of epigenetics-related services, including advanced sequencing technologies to study chromatin accessibility, protein-DNA interactions, and transcription factor binding.
The foundation of ATAC-seq lies in the utilization of the Tn5 transposase. This enzyme is adept at cleaving chromatin and inserting adapter sequences, with a noticeable preference for open chromatin regions. This predilection results in a higher frequency of adapter insertion within accessible chromatin compared to condensed regions. Specifically, Tn5 transposase binds to DNA in these open areas, cleaves it, and inserts predetermined adapter sequences at the cut sites. The locations of these insertions are then amplified via PCR and analyzed using high-throughput sequencing technologies, ultimately revealing the positions of accessible chromatin regions.
The methodology of ATAC-seq is notably straightforward, negating the need for prior selection of specific transcription factors or chromatin modifications. As such, it is widely applicable for analyzing chromatin accessibility across various genomes. When compared to traditional techniques for examining chromatin accessibility, such as DNase-seq and MNase-seq, ATAC-seq not only streamlines the operational workflow but also significantly reduces the requirement for sample quantities. This positions ATAC-seq as an instrumental tool in investigating intricate epigenetic phenomena, including gene regulation, transcription factor binding, and chromatin remodeling.
ATAC-seq technology was initially introduced by Buenrostro et al. in 2013, with the objective of providing a more efficient and straightforward tool for genomic and epigenomic research. Prior to the advent of ATAC-seq, researchers predominantly employed techniques such as DNase-seq and MNase-seq to assess chromatin accessibility. However, these methods required large cell samples and involved relatively complex experimental procedures. The introduction of ATAC-seq revolutionized this landscape by enabling the capture of chromatin accessibility information with as few as 500 to 50,000 cells.
Since its inception in 2013, ATAC-seq has been widely applied in epigenetics, genomics, and biomedical research. Particularly between 2013 and 2021, there was an exponential increase in scientific publications related to ATAC-seq, with over 1,000 articles published across diverse fields. The research domains span from fundamental studies of chromatin architecture to the epigenetic mechanisms involved in clinical conditions such as cancer and diabetes. The most prolific journals publishing ATAC-seq studies include Nature Communications and Scientific Reports. Other notable journals include Nucleic Acids Research, Genome Biology, eLife, Cell Reports, Genome Research, Methods in Molecular Biology, and Bioinformatics.
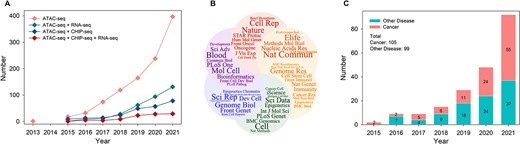 Bibliometrics and datasets statistics of ATAC-seq. (Liheng Luo, et al., Briefings in Bioinformatics, 2022)
Bibliometrics and datasets statistics of ATAC-seq. (Liheng Luo, et al., Briefings in Bioinformatics, 2022)
During this period, ATAC-seq not only achieved significant milestones in basic scientific research but also opened new avenues for clinical therapeutics and disease diagnostics. Its widespread application underscores ATAC-seq's pivotal role in advancing our understanding within the field of epigenetics.
ATAC-seq involves several key steps to capture and analyze chromatin accessibility. These stages help researchers identify open chromatin regions in the genome.
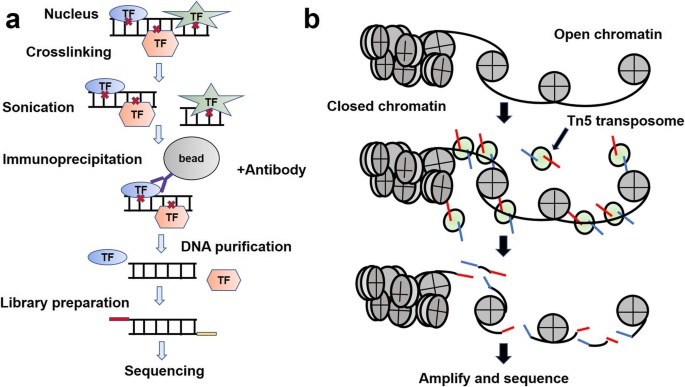 Workflows of ATAC-Seq. (Ma, S., et al., Mol Biomed, 2020).
Workflows of ATAC-Seq. (Ma, S., et al., Mol Biomed, 2020).
Below is a detailed overview of the ATAC-seq experimental procedure:
1. Nuclei Preparation
The initial step involves the extraction of nuclei from the target cells. To ensure the precision and effectiveness of the experiment, cells are lysed using a buffer, allowing for the removal of cell membranes while preserving the nuclei. This step is vital, as only chromatin within intact nuclei can be utilized for subsequent analyses. Typically, gentle lysis is achieved using low-salt buffers or surfactants to maintain nuclear integrity.
2. Fragmentation and DNA Extraction
Following nuclei isolation, chromatin is fragmented using the Tn5 transposase. This enzyme cleaves the chromatin into smaller fragments, targeting regions of open chromatin where it inserts adapter sequences. This process lies at the core of ATAC-seq, as the frequency of transposase insertion is higher in accessible regions than in condensed ones. Subsequently, fragmented DNA is extracted and purified to eliminate impurities, ensuring that high-quality samples are available for further experimentation.
3. PCR Amplification
To obtain sufficient material for sequencing from trace amounts of DNA, the extracted DNA undergoes amplification via Polymerase Chain Reaction (PCR). During this phase, specific adapter sequences ligate with transposed DNA fragments, enabling the generation of vast quantities of templates via PCR amplification. This step not only increases the signal but also ensures an adequate amount of DNA for sequencing.
4. Library Purification
Post-amplification, DNA fragments are subjected to library purification to remove undesirable short fragments, adapter dimers, and potential contaminants. The purified library thus provides a more accurate representation of chromatin accessibility. Techniques like magnetic bead purification or column chromatography are employed to prepare DNA fragments for subsequent sequencing analysis.
5. High-Throughput Sequencing
The purified DNA library is then sequenced using high-throughput platforms such as Illumina. These platforms proficiently sequence vast numbers of DNA fragments, furnishing researchers with extensive data on chromatin accessibility regions. The sequencing output enables further analysis of chromatin openness at various genomic loci, aiding the functional mapping of these areas.
6. Data Analysis
The voluminous data generated from high-throughput sequencing require processing and analysis through specialized bioinformatics tools. Initially, sequencing reads are aligned and mapped to a reference genome to determine insertion site locations. Subsequent statistical analyses facilitate the identification of open chromatin regions across the genome and the construction of chromatin accessibility profiles. Further analyses can elucidate how specific transcription factors or regulatory elements influence chromatin openness under varying conditions, revealing their roles in gene regulation.
ATAC-seq has emerged as a pivotal technology in the realms of epigenomics, gene regulation, and disease research due to its efficiency, simplicity, and ability to derive precise chromatin accessibility data from a minimal number of cells. Here, we outline the principal applications of ATAC-seq:
1. Mapping Chromatin Accessibility and the Epigenomic Landscape: One of the primary utilizations of ATAC-seq lies in mapping chromatin accessibility. By analyzing open chromatin regions across diverse cell types and biological states, researchers can construct comprehensive epigenomic maps. These maps reveal the genomic areas that are open under specific conditions, which is crucial for elucidating the mechanisms governing gene expression regulation. Such insights enable the identification of genomic regions undergoing accessibility changes during processes like cell differentiation, development, stress response, or disease onset.
2. Identifying Key Transcription Factors in Biological Processes: Beyond mere analysis of chromatin accessibility, ATAC-seq facilitates the identification of transcription factors critical for gene expression regulation. By scrutinizing open chromatin regions and integrating information on transcription factor binding sites, researchers can pinpoint key transcription factors involved in particular biological processes or signaling pathways. For example, ATAC-seq can uncover dysregulated transcription factors in cancer cells, advancing our understanding of cancer pathogenesis.
3. Identifying Genes and Targets Regulated by Transcription Factors: When combined with other techniques, ATAC-seq aids in discovering target genes regulated by transcription factors. Through footprinting analysis of transcription factor binding regions, researchers can determine which genes are directly controlled by specific transcription factors. This is essential for unraveling the mechanisms of transcription factor action and their roles in various diseases.
4. Analyzing Chromatin Accessibility Across Different Tissues or Conditions: ATAC-seq unveils variations in chromatin accessibility across different tissues or under various treatment conditions. In disease models, changes in chromatin accessibility may indicate underlying pathogenic mechanisms. By comparing chromatin accessibility maps from different tissues or disease states, researchers can identify disease-related regulatory elements and crucial transcription factors.
5. Studying Nucleosome Positioning: ATAC-seq can also be employed to examine nucleosome positioning. The distribution and structure of nucleosomes in different chromatin regions play a significant role in gene transcription regulation. By exploring the relationship between nucleosome positioning and chromatin accessibility, ATAC-seq aids in elucidating gene regulatory mechanisms, particularly those associated with chromatin remodeling and epigenetic modifications.
6. Generating Features of Transcription Factor Binding Regions (Footprinting): ATAC-seq data allows for footprinting analysis to characterize transcription factor binding regions. This analytical method can identify binding sites of transcription factors on DNA, assisting researchers in understanding how specific transcription factors function in gene regulation. This technique has played a vital role in exploring gene regulatory networks and advancing epigenomic research.
7. Exploring Gene Regulatory Networks and Disease Mechanisms: ATAC-seq is extensively utilized in various mechanistic studies, including exploring signaling pathways, phenotypic changes, and gene regulation in disease contexts. The technology not only reveals the activity of regulatory genes but also, when combined with RNA-seq, CUT&Tag, and other techniques, elucidates changes in regulatory factors during distinct biological processes or disease conditions. For instance, in cancer research, ATAC-seq has helped identify chromatin regions whose openness is associated with tumorigenesis, providing novel targets for early diagnosis and therapeutic intervention.
Although ATAC-seq is widely utilized for its efficiency and ability to generate data on chromatin accessibility from small cell quantities, it presents several inherent limitations, particularly concerning experimental design and data analysis. Here are some of the primary limitations associated with ATAC-seq:
1. Random Junction Ligation Bias
The fundamental mechanism of ATAC-seq involves the insertion of adapter sequences into accessible chromatin regions by the Tn5 transposase. This process, however, is subject to randomness since there is a 50% likelihood that both ends of the same DNA fragment receive identical adapters. Only fragments with different adapters on either end are amenable to enrichment and amplification, leading to a potential loss of data and reduced sequencing coverage and accuracy.
2. Limitations in PCR Amplification of Large Fragments
ATAC-seq can encounter challenges when dealing with overly large DNA fragments, which may impede effective PCR amplification. PCR requires DNA fragments to be within a specific size range for optimal amplification efficiency. Larger fragments might result in data loss or incomplete data, posing a challenge in obtaining sufficient amplifiable DNA fragments under certain cell types or experimental conditions.
3. Dependency on Tn5 Transposase Reaction Conditions
The activity of the Tn5 transposase is highly contingent upon the composition of the reaction solution and the specific experimental conditions. Suboptimal conditions may lead to insufficient transposase activity, consequently affecting the efficiency of chromatin region cleavage. Despite recent advancements, it remains essential to optimize these conditions according to sample type and experimental demands to ensure targeted cleavage and efficient capture of chromatin accessible regions.
4. Challenges in Plant Cell Applications
The implementation of ATAC-seq in plant cells poses distinct challenges due to the presence of a rigid cell wall, which complicates chromatin extraction and handling. Additionally, plant cells contain organelles such as chloroplasts and mitochondria, which could potentially contaminate chromatin samples and compromise result accuracy. Moreover, the lack of stable genetic cell lines in plants necessitates further refinement and optimization for effective ATAC-seq application.
5. Complexity Across Cell Types
When applied to complex cell types, such as multicellular tissues, ATAC-seq may encounter technical difficulties. Variability in chromatin accessibility across different cell types might introduce data heterogeneity and analytical challenges. Although single-cell ATAC-seq technologies have been developed, they still face issues related to cell sorting and data interpretation, potentially affecting the precision and interpretability of experimental results.
ATAC-seq and ChIP-seq are pivotal techniques in epigenetic research. Both are essential for studying chromatin structure and gene regulation, yet they differ markedly in their principles, applications, and objectives.
Comparison of ATAC-seq and ChIP-seq
| Feature | ChIP-seq | ATAC-seq |
| Research Goal | Investigates protein-DNA interactions, especially transcription factor binding sites | Studies chromatin accessibility, marking open chromatin regions |
| Principle | Uses antibodies to capture target protein-DNA complexes, sequencing reveals binding sites | Employs Tn5 transposase to insert adaptor sequences marking open chromatin |
| Technical Dependency | Relies on immunoprecipitation and protein-DNA crosslinking | Depends on transposase's recognition and marking of open chromatin |
| Applications | Identifies transcription factor binding sites, elucidates regulatory roles | Maps genome-wide chromatin accessibility, infers potential regulatory factors |
| Sample Requirement | Requires a large number of cells, typically targeting specific transcription factors | Low sample requirement, simple procedure suitable for large-scale genome analysis |
| Scope | Studies transcription factor target genes and epigenetic modification mechanisms | Analyzes gene regulatory networks, signaling pathways, and chromatin accessibility |
| Advantages | High specificity for precise localisation of transcription factor binding sites | Efficient, requires fewer cells, capable of mapping open chromatin landscapes |
Is Combined Use Necessary?
While ATAC-seq and ChIP-seq each have distinct use cases, their combination often yields more comprehensive biological insights. By integrating ATAC-seq and ChIP-seq, researchers can first map open chromatin regions using ATAC-seq and then employ ChIP-seq to precisely pinpoint transcription factors or other regulatory proteins binding within these regions. This dual approach elucidates the dynamic interplay between chromatin accessibility and gene expression regulation.
Additionally, coupling ATAC-seq with RNA-seq provides another synergistic avenue. Here, ATAC-seq identifies changes in chromatin accessibility, while RNA-seq assesses the expression of genes associated with these regions. Such integration helps uncover the underlying causes of gene expression changes and the relationships between chromatin state and transcriptional outputs.
Common Combinatorial Approaches
1. ATAC-seq + RNA-seq: This combination allows researchers to obtain insights into chromatin accessibility changes, particularly at gene promoters, followed by differential gene expression analysis through RNA-seq. This approach helps elucidate the comprehensive regulatory architecture and identify potential regulatory factors.
2. ATAC-seq + ChIP-seq + RNA-seq: This multi-faceted approach offers a robust framework for understanding chromatin accessibility, transcription factor binding, and gene expression regulation. ATAC-seq identifies open chromatin regions, ChIP-seq verifies and accurately locates transcription factor binding sites, and RNA-seq evaluates changes in gene expression. Such detailed analyses can illuminate the connections between chromatin accessibility, transcription factor interaction, and downstream target gene regulation, providing powerful insights into gene regulatory networks.
If you want to learn more about ATAC-seq, you can refer to the following article:
ATAC-Seq – A Method to Study Open Chromatin
How to Interpret ATAC-Seq Data
Quality Control of ATAC Sequencing Library
ChIP-seq vs. ATAC-seq
References:


