
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

Chromosomes are structures formed through the extensive condensation and spiraling of chromatin. Similar to compressed files in computing, this condensed state is not conducive to reading (transcription). Consequently, prior to transcription for protein synthesis, chromatin must be decompressed or opened to render the DNA readable.
Among various epigenetic technologies, Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) has emerged as a prominent method for assessing chromatin accessibility across the entire genome. This technology directly evaluates the "readability" of chromatin, highlighting its significant application value. Despite its importance, many online resources about ATAC-seq primarily focus on analytical methods and workflows, with limited discussion on the biological implications of the results. This review aims to address the following aspects:
1. Alignment and Sequencing Quality
2. Peak Calling
3. Downstream Analysis
4. Integration with Multiomics Data
5. Visualisation
In alignment with the analytical workflow for all next-generation sequencing technologies, the initial step in ATAC-seq analysis involves mapping short sequence reads to a reference genome, followed by preliminary quality control measures.
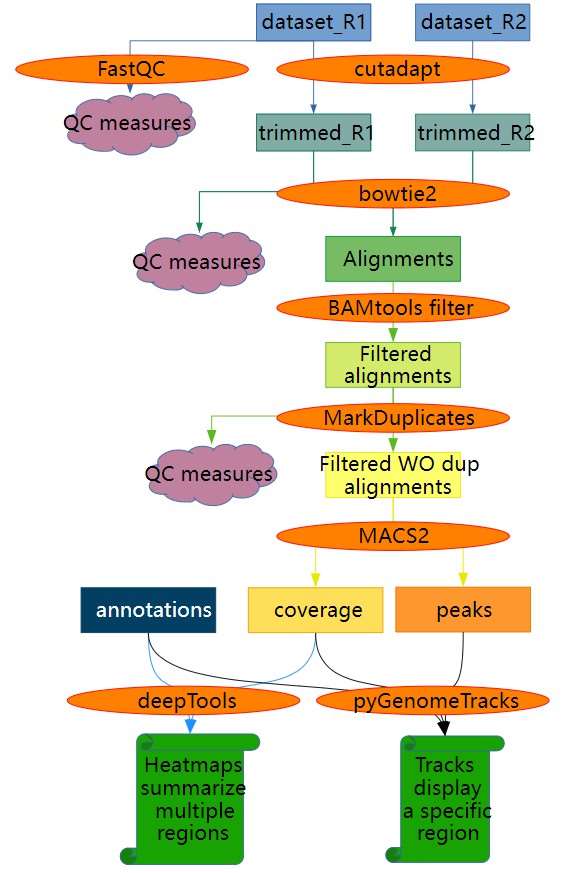 Figure 1: Overview of ATAC-Seq data analysis. (From galaxyproject.org)
Figure 1: Overview of ATAC-Seq data analysis. (From galaxyproject.org)
The diagram above illustrates that each step in the analysis process (indicated by red arrows) involves quality control measures. Quality control is critical for obtaining accurate analytical results. In ATAC-seq, quality control primarily focuses on analyzing insertion tags within libraries and transcription start site (TSS) signals. This can be understood through the following details:
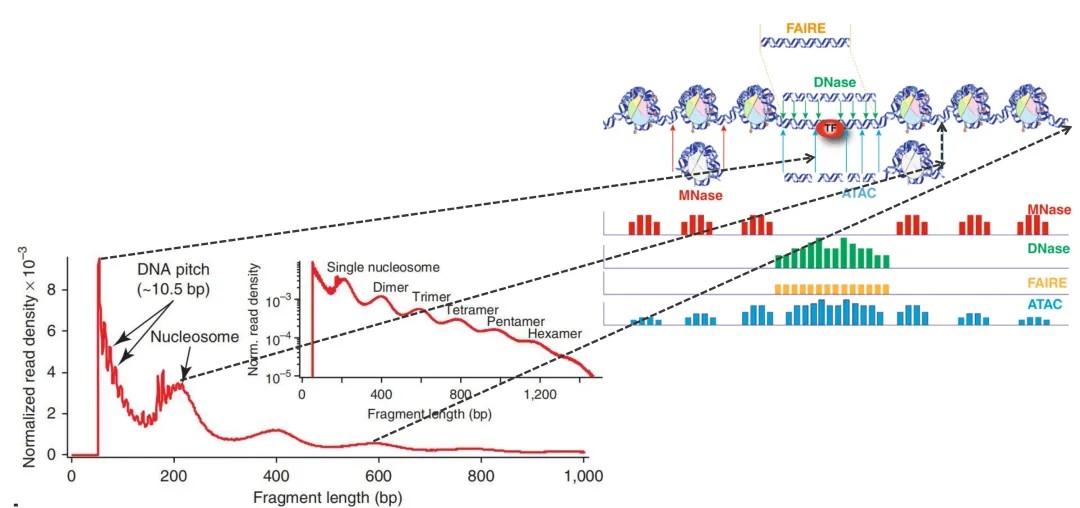 Figure 2: Quality control analysis showing peak distribution and nucleosome cleavage patterns. (Kevin W. Trotter 2011)
Figure 2: Quality control analysis showing peak distribution and nucleosome cleavage patterns. (Kevin W. Trotter 2011)
The first peak observed before 150 base pairs represents the cleavage of open chromatin regions. This peak is accompanied by periodic, sawtooth-like small peaks, with approximately 10 base pairs between each minor peak. The peak around 200 base pairs is primarily attributed to the cleavage of nucleosomes. As previously mentioned, DNA wrapped around a nucleosome is approximately 147 base pairs long; due to variability in cleavage precision, the observed peak occurs near 200 base pairs. Subsequent peaks represent the presence of two, three, or multiple nucleosomes, with decreasing peak heights. This reduction indicates a lower probability of cutting nucleosomes located further from the chromatin's proximal region.
The peaks identified by ATAC-seq and ChIP-seq represent different biological phenomena and therefore have distinct meanings:
ChIP-seq involves the use of antibodies specific to a target protein to precipitate the protein and associated DNA fragments. These DNA fragments are subsequently mapped to the genome. The binding sites of the target protein are indicated by regions where DNA fragments are densely stacked. Visualization of these regions as bar graphs results in discrete peaks. Typically, ChIP-seq yields a single prominent peak corresponding to the binding site of the protein of interest.
ATAC-seq relies on the Tn5 transposase to cleave accessible DNA sites within chromatin. The binding of Tn5 transposase to chromatin is a stochastic event. The determination of whether a location's read depth constitutes a peak is performed using software such as MACS (Model-based Analysis of ChIP-Seq). When a transcription factor binds to DNA, it obstructs the Tn5 transposase from cutting at that specific site, resulting in a protective region where reads are depleted. Consequently, the regions bound by transcription factors in ATAC-seq typically exhibit a characteristic valley-like peak.
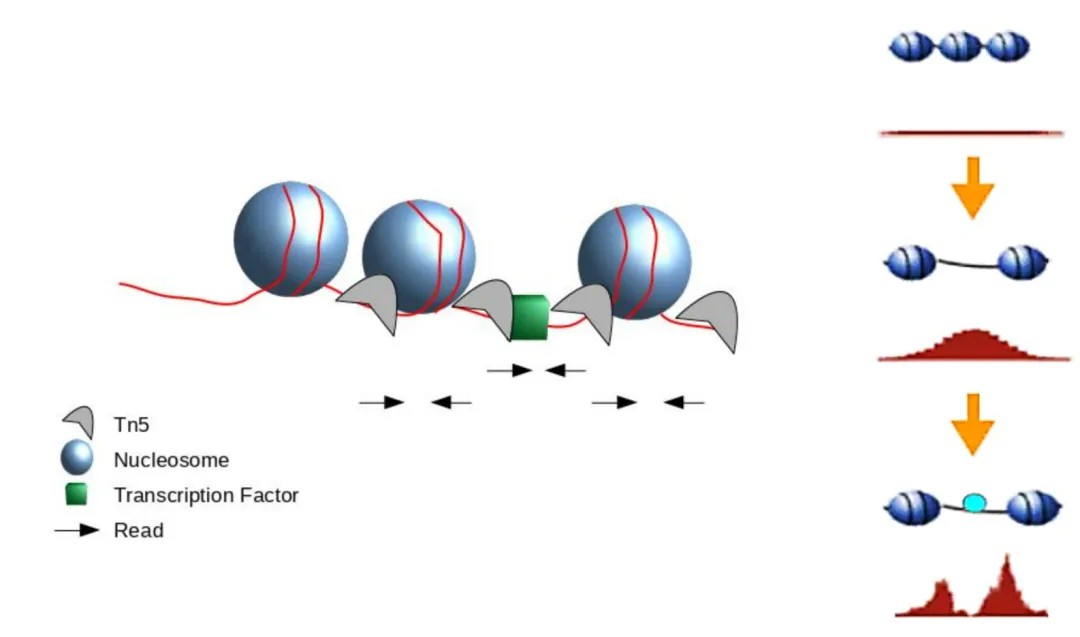 Figure 3. Scheme of ATAC-Seq reads relative to nucleosomes. (Kia et al. 2017.)
Figure 3. Scheme of ATAC-Seq reads relative to nucleosomes. (Kia et al. 2017.)
When employing MACS software for peak calling, the methodology used to construct the model significantly influences the criteria MACS employs to identify peaks.
MACS utilizes a statistical model to distinguish between true signal peaks and background noise. The parameters set during model construction, including the choice of input control, the peak enrichment threshold, and the model's sensitivity, directly affect the accuracy and reliability of peak detection. Variations in these parameters can lead to differences in the number and characteristics of detected peaks, emphasizing the importance of carefully calibrating the model to reflect the experimental conditions and goals.
By optimizing these parameters, researchers can enhance the specificity and sensitivity of peak detection, thereby improving the interpretability and biological relevance of the results obtained from ATAC-seq and similar high-throughput sequencing techniques.
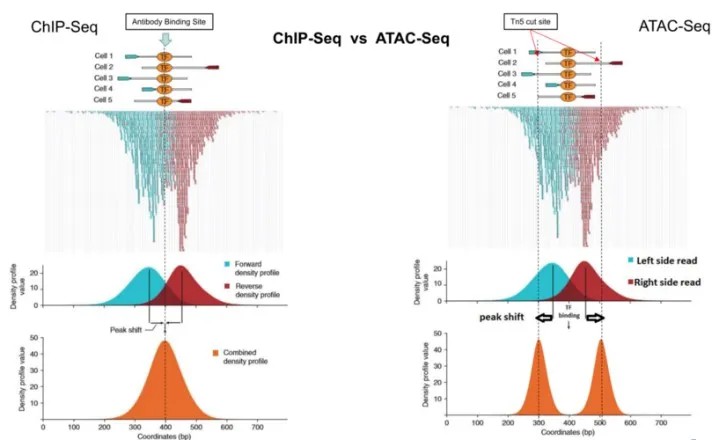 Figure 4. Integration of ATAC-seq with Other Sequencing Technologies
Figure 4. Integration of ATAC-seq with Other Sequencing Technologies
Both ChIP-seq and ATAC-seq generate distinct read binding patterns that can manifest as double peaks at regions of transcription factor (TF) or Tn5 integration.
In ChIP-seq, the observed peaks reflect the regions where DNA fragments co-precipitated with TFs are found. However, these peaks often extend beyond the actual TF binding sites due to the inclusion of surrounding DNA fragments, necessitating an inward shift of the read positions to accurately represent the TF binding sites.
Conversely, in ATAC-seq, a shift is also required to align adjacent peaks into a single peak; this shift should be directed outward from the center of the peak. This adjustment accounts for the enrichment of reads at the flanking regions of the TF binding sites rather than at the central motif.
For instance, in the case of CTCF (CCCTC-binding factor), ChIP-seq peaks delineate the CTCF binding regions, with the central location representing the CTCF motif. In contrast, ATAC-seq reads are enriched at the flanking regions of the motif, as illustrated in the accompanying figure. The horizontal axis of the figure represents genomic coordinates, while the vertical axis denotes the signal intensity of ATAC-seq.
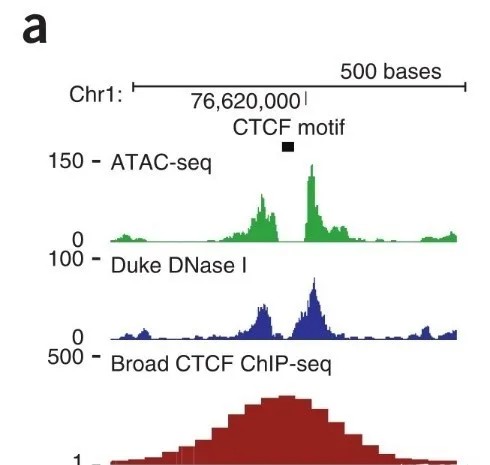 Figure 5. Illustration of CCCTC-Binding Factor (CTCF)
Figure 5. Illustration of CCCTC-Binding Factor (CTCF)
Downstream analysis represents a primary focus of this article, and it is categorized into four main aspects: peak analysis, motif analysis, nucleosome positioning, and TF footprinting.
Peak analysis is subdivided into two principal approaches:
Differential Peak Analysis Based on Predefined Peak Sets: This method involves identifying peaks from a predefined set and subsequently performing differential analysis based on RNA sequencing (RNA-seq) data or other analogous methods. It is recommended that all samples be pooled together to identify an unbiased and consistent set of peaks before conducting further analyses.
Sliding Window Approach: This method operates with fewer assumptions, thus offering a more unbiased analysis. However, it is noted that this approach may result in a higher false positive rate, necessitating more stringent preliminary filtering.
Peak annotation involves mapping peaks to functional genomic regions—such as exons, promoters, enhancers, and untranslated regions (UTRs)—to elucidate the regulatory functions of peaks on genes. Typical ATAC-seq peaks generally fall within cis-regulatory elements. Following peak annotation, functional enrichment analysis can be conducted on the resulting gene list to further explore the biological significance of the identified peaks.
TFs typically influence transcription by binding to motifs located within open chromatin regions. Thus, the analysis of motifs and TF binding sites constitutes a crucial component of ATAC-seq analysis. The human genome encompasses approximately 1,600 distinct transcription factors, with their binding sites dispersed throughout the genome. Analyzing the activity and accessibility of various motifs facilitates the identification and interpretation of key biological regulatory processes.
Annotation: Numerous databases provide experimentally or algorithmically predicted motifs for annotation purposes, including CIS-BP and RegulonDB. These resources are instrumental in motif annotation.
Enrichment: Upon identifying motifs, calculating their frequency within open peak regions allows for the detection of enriched motifs, which can subsequently be used to predict or associate TF activity.
An alternative approach to describing TF regulation involves the use of footprints. Active TF binding results in the failure of Tn5 transposase to bind during the ATAC-seq library preparation, leading to the formation of a dip (or "footprint") at the TF binding site within the peak. It is noteworthy that accurate footprint identification is challenging and relies on high sequencing depth. Additionally, many algorithms currently used for TF footprint analysis were not specifically developed for ATAC-seq, which may introduce biases into the results.
In typical ATAC-seq datasets, longer fragments often represent regions associated with multiple nucleosomes. Several methods exploit this information to detect nucleosome-enriched regions. However, due to the lower read coverage in these regions compared to open chromatin areas, such analyses can be particularly challenging.
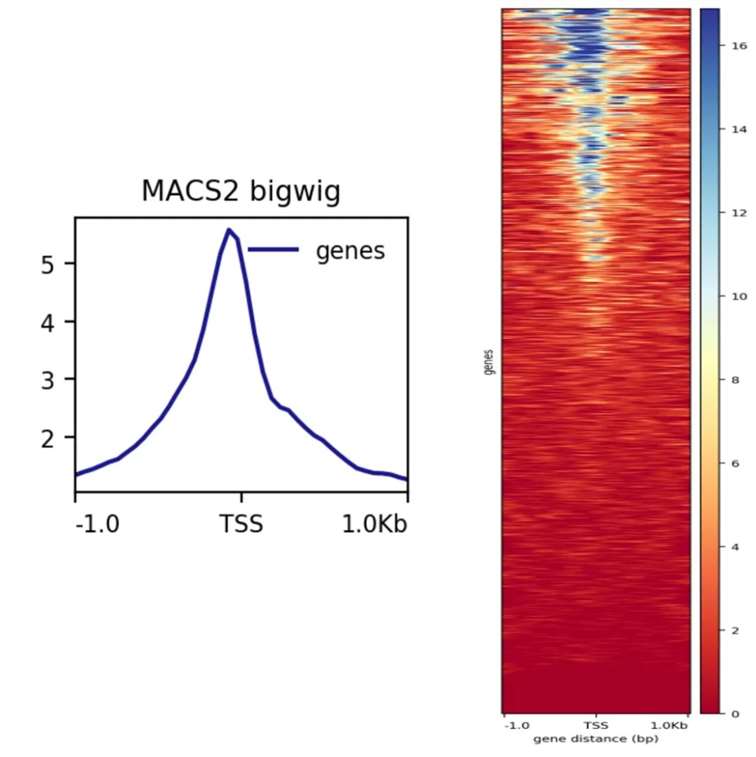
Visualization of genomic data is frequently accomplished through the depiction of peaks and heatmaps centered on TSS. Such visualizations are pivotal for interpreting chromatin accessibility and identifying regulatory elements.
The graphical representation typically involves plotting peaks around the TSS, with each line in the plot representing a distinct transcript. These visual tools enable the identification of open chromatin regions, potential enhancers, or silencers.
The promoter regions are often delineated within a commonly used range of 2.5 kilobases (kb) from the TSS. Since promoter regions lack a well-defined boundary, this range provides a practical approximation for their identification.
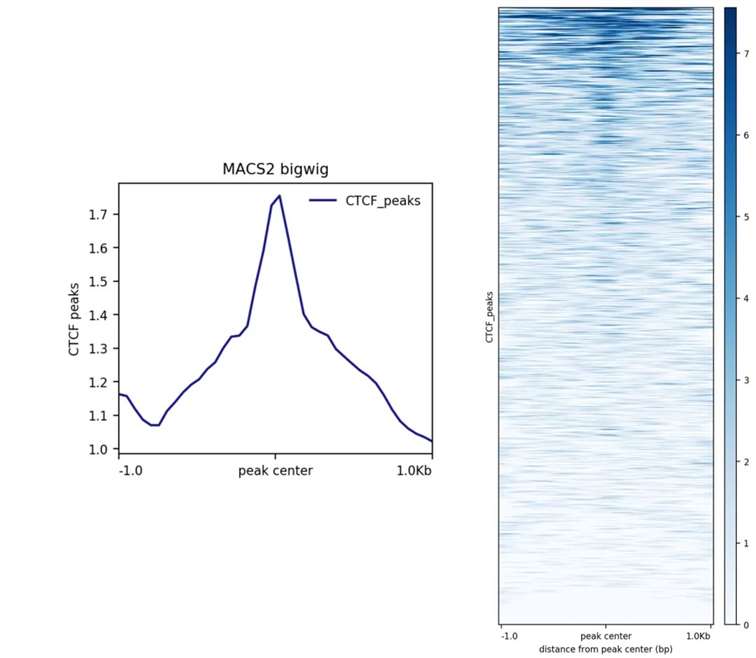
In addition to visualizing data centered on TSS, graphical representations can also be centered on specific gene peaks, as illustrated below:
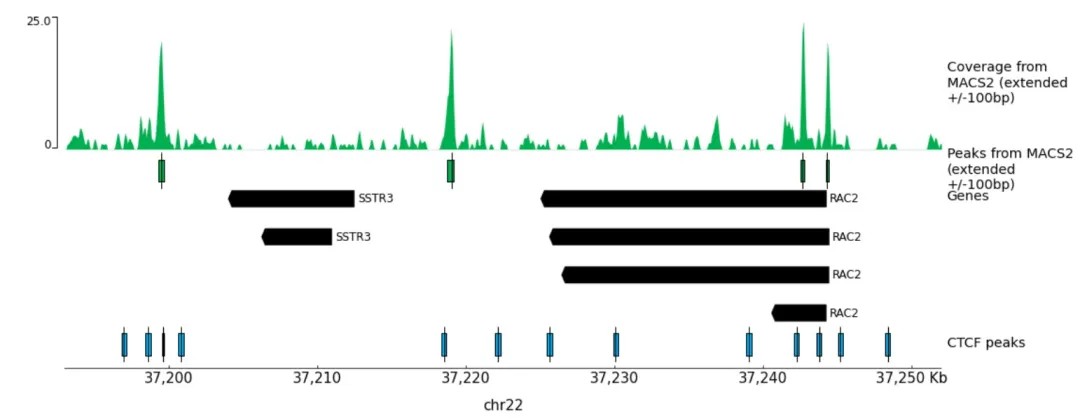
Additionally, it is common to visualize specific DNA regions within articles, such as the RAC2 gene illustrated below:
This version maintains a formal and precise tone, suitable for scientific communication.
The question of whether ATAC-seq alone can replace ChIP-seq is addressed negatively. In practice, ATAC-seq is frequently combined with other sequencing technologies to achieve comprehensive analyses.
Typically, RNA-seq is conducted prior to ATAC-seq. Differentially expressed genes identified through RNA-seq can be further investigated using ATAC-seq to perform motif analysis, which aids in identifying regulatory factors associated with target genes. Subsequent experimental validation can then be conducted to confirm these findings.
Alternatively, ATAC-seq can be utilized to examine chromatin accessibility, with the goal of determining whether changes in chromatin state correlate with increased transcript levels. This approach allows RNA-seq to identify genes corresponding to enriched transcripts, facilitating functional analysis of these genes and integration with phenotypic validation. This creates a comprehensive framework encompassing epigenetic regulation, expression, function, and phenotype.
ChIP-seq is often employed following ATAC-seq to provide additional validation. For instance, after identifying peaks with ATAC-seq and detecting motifs associated with specific transcription factors, ChIP-seq can be utilized to pinpoint the binding sites of these transcription factors. This approach enables the determination of whether the transcription factors are interacting with promoter regions or enhancer regions.
Additionally, the advent of single-cell RNA-seq has led to the development of emerging techniques such as scATAC-seq combined with scRNA-seq, which allows for the examination of chromatin accessibility at the single-cell level.
ATAC-seq enables the identification of open regions for TF motifs across the entire genome, facilitating the discovery of regulatory elements such as enhancers that may be located at significant distances from their target genes. This capability is crucial for constructing complex regulatory networks, including enhancer-promoter interactions.
References:


