Chromatin regulatory regions play pivotal roles in numerous disease processes and embryonic development. To unravel the genomic regulatory landscape of cells and tissues, cutting-edge epigenomic sequencing technologies like Chromatin Immunoprecipitation Sequencing (ChIP-seq) and Transposase-Accessible Chromatin High-Throughput Sequencing (ATAC-seq) have emerged. These techniques allow us to explore specific chromatin states and their associated transcription factors, providing valuable insights into the temporal and spatial dimensions of gene regulation.
The advent of Chromatin Immunoprecipitation Chip (ChIP-chip) technology has further expanded the repertoire of epigenomic analysis methods. Notable techniques include ChIP-seq, DNase I Ultrasensitive Site Sequencing (DNase-seq), and ATAC-seq. These powerful approaches enable researchers to comprehensively investigate the dynamic interactions between DNA and proteins, shedding light on the complex regulatory networks that govern cellular processes and influence disease outcomes.
ChIP-based Methods
Advancements in ChIP-based research methods have brought forth a more powerful and refined approach compared to microarrays. ChIP-seq offers several key advantages, including high resolution, reduced noise, and extensive coverage. As the cost of second-generation sequencing continues to decline rapidly, CHIP-seq is poised to become an indispensable tool in the study of gene regulation and epigenetics.
In contrast to microarrays, ChIP-seq provides a comprehensive and detailed view of DNA-protein interactions, allowing researchers to better characterize sequence motifs and identify critical transcription factors, enhancers, and other regulatory elements. This enhanced precision and sensitivity make ChIP-seq an invaluable asset in unraveling the complexities of genomic regulation and shedding light on the mechanisms underlying various biological processes.
Please refer to our article ChIP-Seq: A Versatile Tool for Epigenomics.
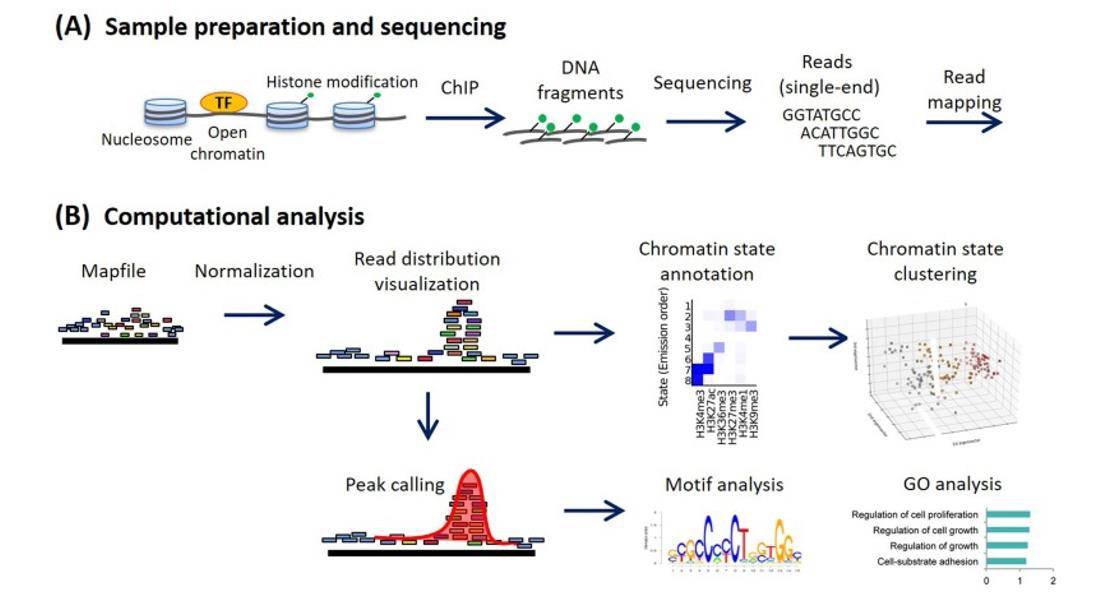 Methods for ChIP-seq analysis. (Nakato et al., 2021)
Methods for ChIP-seq analysis. (Nakato et al., 2021)
Traditional ChIP-Seq
The traditional ChIP-seq technique, a cornerstone in genomics research, aims to capture DNA bound to specific proteins. This multi-step process involves crosslinking DNA and proteins in situ using formaldehyde, followed by sonication to create small DNA fragments of 200-600 base pairs. Immunoprecipitation with specific antibodies is then performed to isolate DNA-protein complexes, which are subsequently de-crosslinked. The released DNA undergoes library preparation steps, such as end repair, junction ligation, and sequencing.
However, ChIP-seq has certain limitations. Polymerase chain reaction (PCR) bias and limited amplification length can introduce artifacts. GC bias during lysis and sequencing may affect the data quality. Moreover, the requirement for a considerable number of cells (105 ∼107 cells) can result in substantial loss during the immunoprecipitation process. Additionally, the formaldehyde cross-linking process may mask antigenic epitopes of transcription factors, leading to potential false-positive results. An alternative approach, known as natural ChIP (N-ChIP), eliminates the issue of masking epitopes and can be employed when dealing with a very small number of cells.
ChIP technology for Microcells
To overcome the limitation of requiring a large number of cells, researchers have explored ChIP technology for microcells. Various methods have been developed to optimize experiments for low cell numbers. Techniques such as chemical blotting in combination with chromatin immunoprecipitation and Tn5 transposase for sequencing library preparation have enabled histone ChIP-seq using as few as 10,000 cells. Ultra-low-input MNase-based natural CHIP (ULI-NChIP) has generated high-quality histone modification profiles from 103 to 106 embryonic stem cells, thanks to adaptations in the NChIP protocol. Another innovative approach, microfluidic oscillatory washing-based ChIP-seq (MOWChIP-seq), even allows genome-wide analysis of histone modifications using only 100 cells. While these methods can provide relatively accurate histone modification profiles in a few cells, their efficacy is limited for many transcription factors due to the scarcity of binding sites on the genome.
Single-cell ChIP-seq (scChIP-seq)
Enter the realm of single-cell ChIP-seq (scChIP-seq), which offers a groundbreaking perspective. Unlike bulk ChIP-seq which captures chromatin features from a population of cells, scChIP-seq enables researchers to study genetic diversity within heterogeneous cell populations and comprehend the evolutionary dynamics of tumor populations. Droplet single-cell ChIP-seq (Drop-ChIP) integrates a microfluidic device with single-cell DNA, enabling the generation of relatively low coverage maps per cell.
Recommended Reading: The Advantages and Workflow of ChIP-seq.
ATAC-Seq
ATAC-seq (Assay for Transposase-Accessible Chromatin sequencing) technology has revolutionized transposase-open chromatin assays, allowing researchers to simultaneously identify open chromatin regions, nucleosome localization, and regulatory motifs with reduced sample requirements (as low as 500~5000 cells). By utilizing motif inference, scientists have successfully deduced the binding sites of DNA-binding proteins in B-cell lines using ATAC-seq.
Recommended Reading: ATAC-Seq – A Method to Study Open Chromatin.
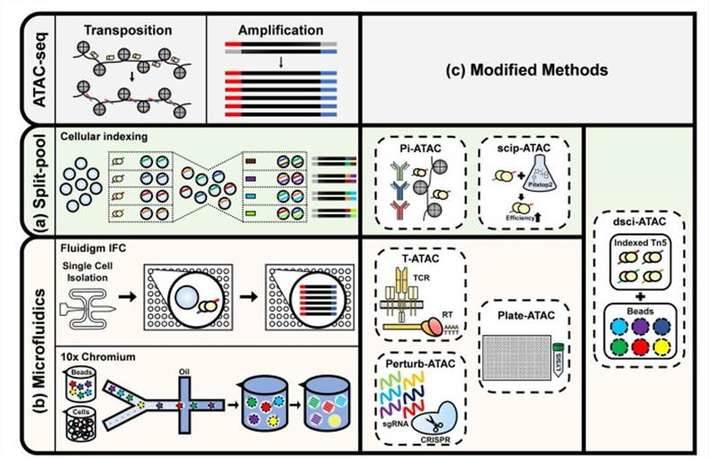 Single-cell ATAC sequencing analysis. (Baek et al., 2020)
Single-cell ATAC sequencing analysis. (Baek et al., 2020)
One of the key advantages of ATAC-seq is its relatively simple and efficient "two-step" library preparation procedure: transposition and PCR. In ATAC-seq experiments, nuclei are isolated and collected, and the chromatin inside the nucleus is fragmented using Tn5 transposase, which then ligates sequencing junctions, streamlining the experimental process significantly. The tightly packed chromatin DNA cannot enter the transposome, whereas chromatin DNA in open regions is randomly inserted and fragmented by the transposome. The resulting fragmented DNA is collected for subsequent analysis.
The most noteworthy innovation of ATAC-seq lies in the use of Tn5 transposase. Initially, Tn5 transposase exhibits low transposition activity, but through targeted modifications, it can be converted into a highly active form with a higher affinity for DNA, making it more amenable for scientific research. By assembling Tn5 transposase in vitro with designed articulators, researchers can form active transposome complexes, enhancing the efficiency and accuracy of the assay.
However, it is crucial to acknowledge that Tn5 transposase does introduce bias due to sequence-dependent binding. Nonetheless, computational tools have been developed to correct this transposition bias, ensuring the accuracy and reliability of the ATAC-seq data.
Combination of ChIP-seq and ATAC-seq
When comparing multiple samples, relying solely on ChIP-seq may pose challenges in identifying significant regulatory elements. In such cases, ATAC-seq emerges as a valuable complement due to its high resolution and signal-to-noise ratio, enabling the identification of differentially regulated sequences, including enhancers. Integrating ChIP-seq with ATAC-seq simplifies the identification of distinct peaks, enhancing the analytical power.
While techniques like DNase-seq and ATAC-seq can infer genomic features, they cannot entirely replace the information provided by ChIP-seq. All these methods indirectly detect transcription factor-DNA binding sites, necessitating further validation of the inferred transcription factor action sites based on enriched motifs.
ChIP-seq directly identifies specific DNA-protein interactions, whereas ATAC-seq reveals genome-wide chromatin openness, providing a unique perspective on the chromatin regulatory landscape. By employing both ATAC-seq and ChIP-seq in combination, researchers can achieve a more comprehensive and profound understanding of chromatin regulation dynamics and its biological implications.
However, there are potential limitations when using ATAC-seq in isolation:
- Not all chromatin regulators possess corresponding motifs; for instance, chromatin remodeling proteins lack specific DNA sequences. Interactions between these proteins and nucleosomes are critical for early embryonic development and cell fate decisions, and their binding cannot be inferred solely from open chromatin information.
- Certain motifs may be bound by multiple sequence-specific transcription factors, leading to ambiguity in transcription factor identification.
- Some homologous transcription factors share similar motif patterns, making it challenging to directly assign open chromatin to a specific individual transcription factor. In such cases, direct detection of interactions between specific transcription factors and DNA remains more reliable.
In summary, the combined usage of ChIP-seq and ATAC-seq offers a powerful approach to gain deeper insights into the regulatory landscape of chromatin. While ATAC-seq complements ChIP-seq by providing a broader chromatin openness perspective, ChIP-seq ensures the direct identification of specific DNA-protein interactions. This integration holds the potential to uncover intricate gene regulation dynamics and unravel the functional significance of chromatin modifications in various biological contexts.
References:
-
Nakato, Ryuichiro, and Toyonori Sakata. "Methods for ChIP-seq analysis: A practical workflow and advanced applications." Methods 187 (2021): 44-53.
- Baek, Seungbyn, and Insuk Lee. "Single-cell ATAC sequencing analysis: From data preprocessing to hypothesis generation." Computational and structural biotechnology journal 18 (2020): 1429-1439.

 Sample Submission Guidelines
Sample Submission Guidelines


