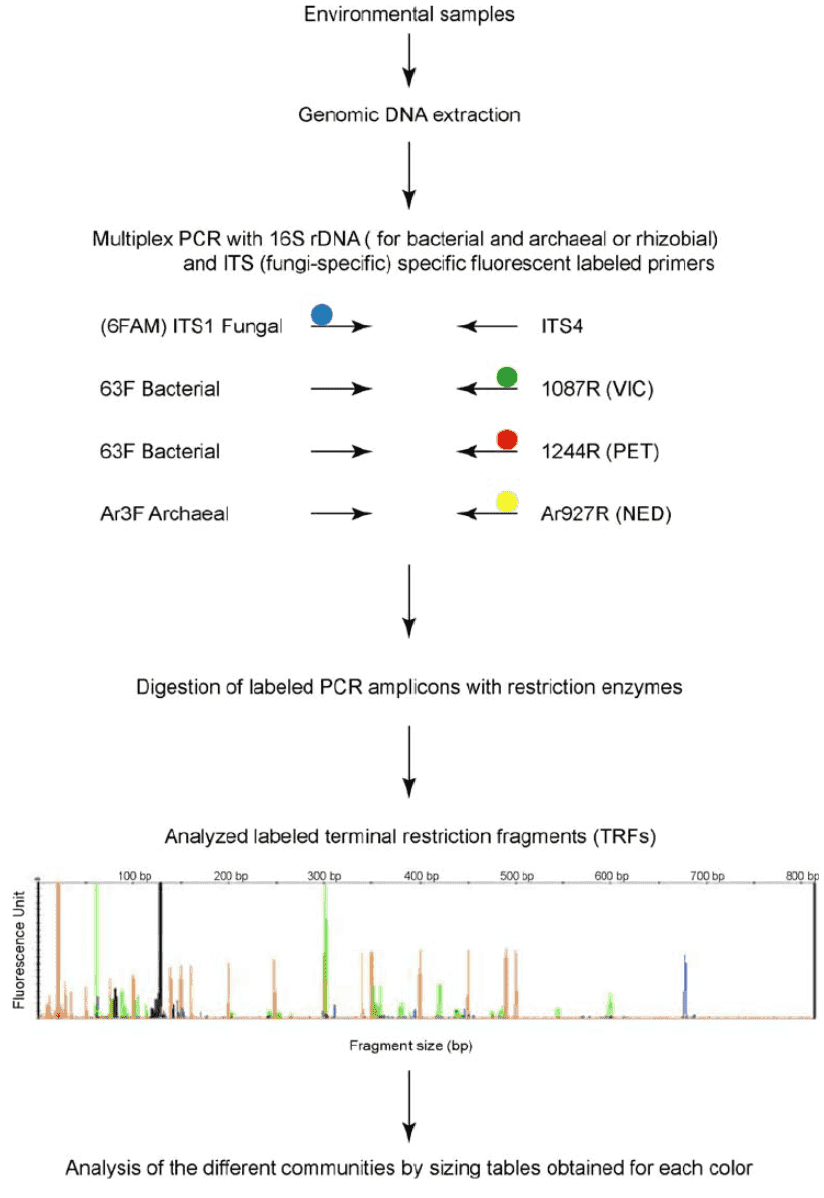
Bacterial Transformation Steps
Preparation of Competent Cells (Part 1): Bacterial Activation and Expansion
How do we obtain competent cells? Specifically, the process begins with the activation of Escherichia coli (E. coli) culture. To achieve this objective, the following steps are undertaken: a small volume of bacterial culture is inoculated into a test tube containing 4 mL of antibiotic-free Luria-Bertani (LB) broth and placed in a shaking incubator at 37°C for overnight incubation, yielding a fresh E. coli culture. Subsequently, the activated culture is inoculated into a centrifuge tube containing 50 mL of antibiotic-free LB broth for scale-up cultivation. Throughout the cultivation process, close monitoring of the culture's status is imperative. Upon the appearance of turbidity in the culture, optical density at 600 nm (OD600) readings should be taken every 20 to 30 minutes. Cultivation should be promptly halted when the OD600 value reaches approximately 0.6.
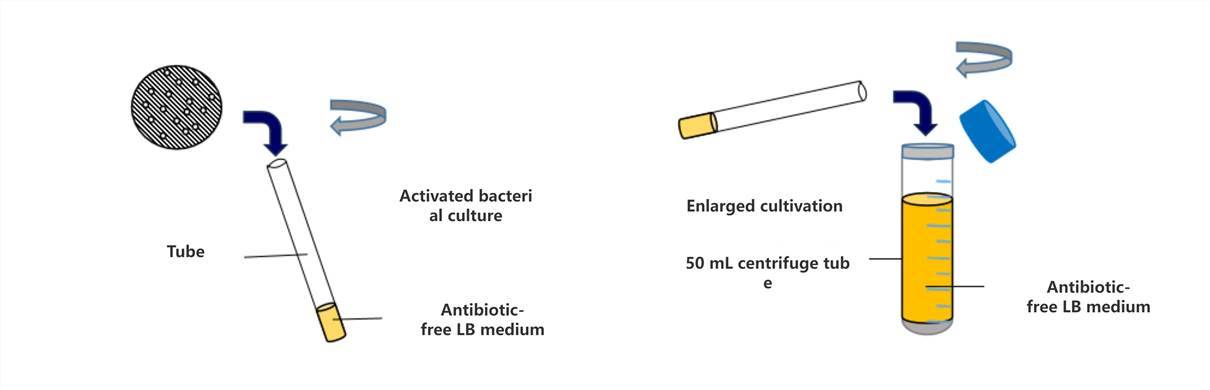
The preserved bacterial strains are resuscitated on antibiotic-free LB agar plates, followed by single colony selection and activation.
Upon completion of activation, scale-up cultivation is performed using large-volume containers such as conical flasks or centrifuge tubes.
Preparation of Competent Cells (Part 2): Preparation
Subsequently, the culture suspension is withdrawn and subjected to a 10-minute ice bath, followed by centrifugation at 5000 revolutions per minute (rpm) for 10 minutes at 4°C. The supernatant is then discarded, and the bacterial cells are gently resuspended in 5 mL of ice-cold CaCl2 solution to ensure uniformity, followed by incubation on ice for 30 minutes. Subsequent centrifugation at 5000 rpm for 10 minutes at 4°C is performed, followed by discarding the supernatant and gentle resuspension of bacterial cells in 5 mL of ice-cold CaCl2 solution to achieve homogeneity. After brief incubation on ice, the competent cell suspension is prepared. It is aliquoted into 1.5 mL centrifuge tubes, preferably utilized promptly for transformation, or alternatively stored for future use under ultra-low temperature conditions at -70°C.
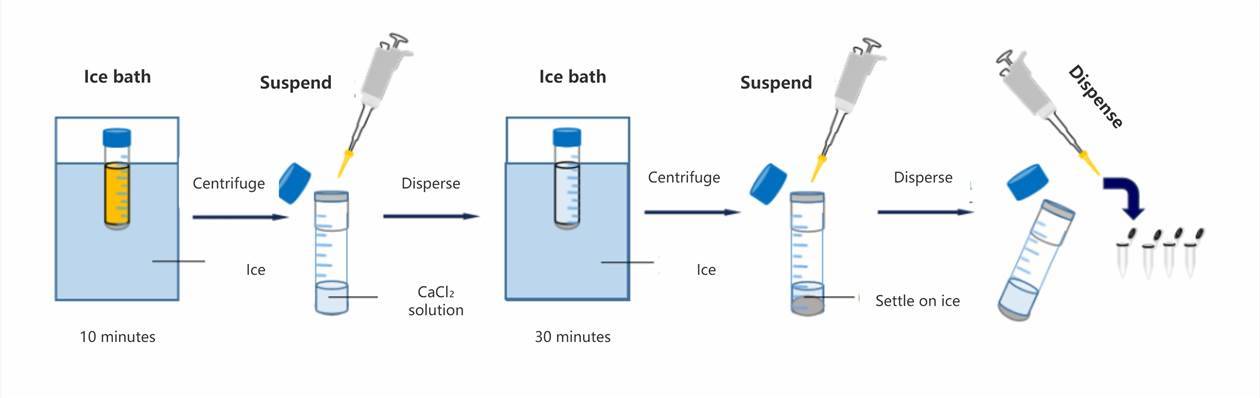
Pre-cool instruments such as centrifuges and reagents like CaCl2 solution before commencing experiments.
Perform all procedures swiftly on ice to prevent warming and contamination.
Preparation of Recombinant Vectors
Subsequently, we delve into the intricacies of the heat shock transformation process. In the standard protocol of bacterial transformation, the construction of vectors stands as the pivotal objective, necessitating the efficient fusion of target genes with the vector. Typically, we opt for restriction enzymes specific to multiple cloning sites on the vector, enabling dual enzyme digestion of both the vector and the target gene fragments. Subsequently, employing T4 ligase, the digested vector and target gene fragments are precisely ligated to attain the desired fusion product. This process demands meticulous manipulation and stringent control to ensure both efficiency and accuracy of the ligation process.
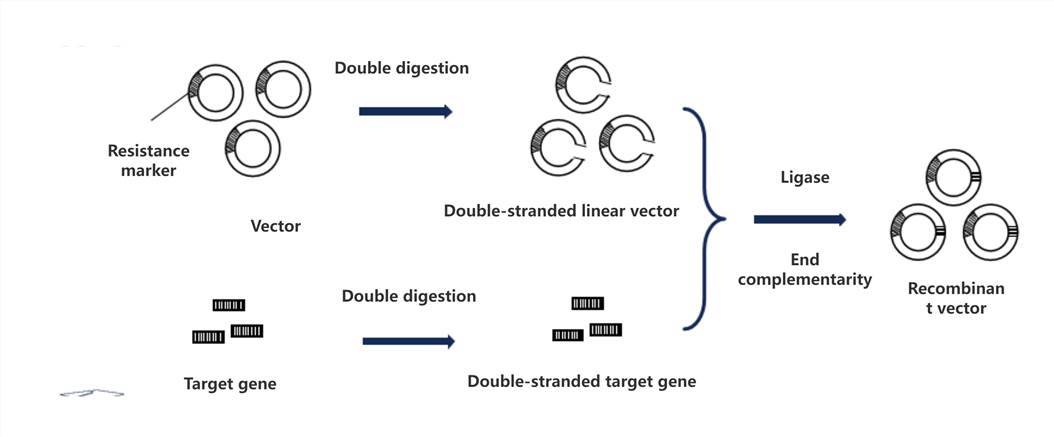
Fragments of the vector are simultaneously cleaved! Cohesive endonucleases incise both the vector and target gene (or compatible ends enzymes).
Complementary ends unite! Ligases facilitate the dual-end complementary joining of the vector and target gene.
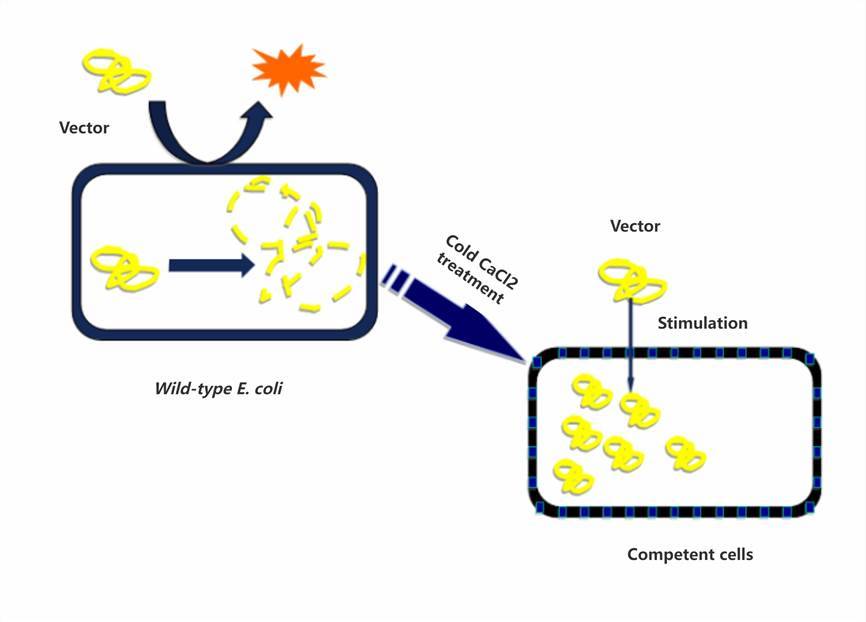
Thermal Induction of Competent Cell Transformation
The transformation mixture, containing the carefully prepared recombinant product, is gently introduced into competent cells, ensuring even distribution. Subsequently, the mixture is incubated on ice for 30 minutes to facilitate cell adhesion to the recombinant product. Following this, the mixture is swiftly transferred to a preheated water bath set to 42°C for a 90-second heat shock, inducing cellular uptake of the recombinant product. Immediately post-heat shock, the mixture is returned to ice and allowed to cool for 2 minutes to prevent thermal damage. Subsequently, 700 microliters of antibiotic-free LB liquid culture medium is added, and the mixture is gently mixed. Finally, the mixture is incubated in a shaking incubator at 37°C for 40 minutes to allow the recipient cells to resume normal growth and facilitate the generation of transformants with the desired resistance characteristics.
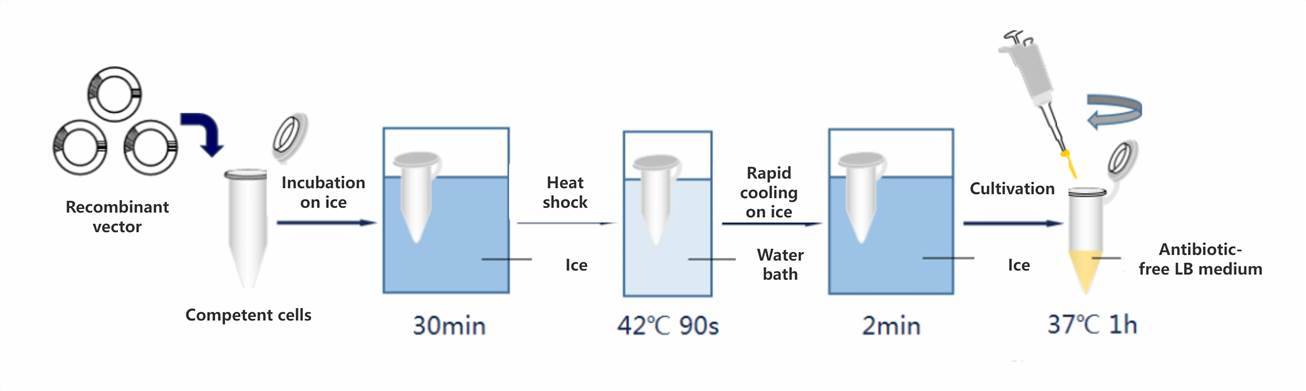
The alternation between cold and heat stimuli induces changes in the permeability of sensory cells.
Upon exposure to heat stimulation, sensory cells become activated, while they persist within the cellular body following the restoration of permeability during cold immersion.
Application of Recombinant Culture Suspension
The cultured bacterial suspension, following amplification, is subjected to centrifugation at a continuous rate of 3000 revolutions per minute for 5 minutes. Subsequently, the majority of the supernatant is removed, leaving approximately 100 microliters for resuspension. This resuspended volume is uniformly spread onto the surface of solid LB agar containing specific antibiotics. Finally, the prepared agar plates are placed in a constant temperature incubator at 37°C and allowed to incubate undisturbed for 12 to 16 hours.
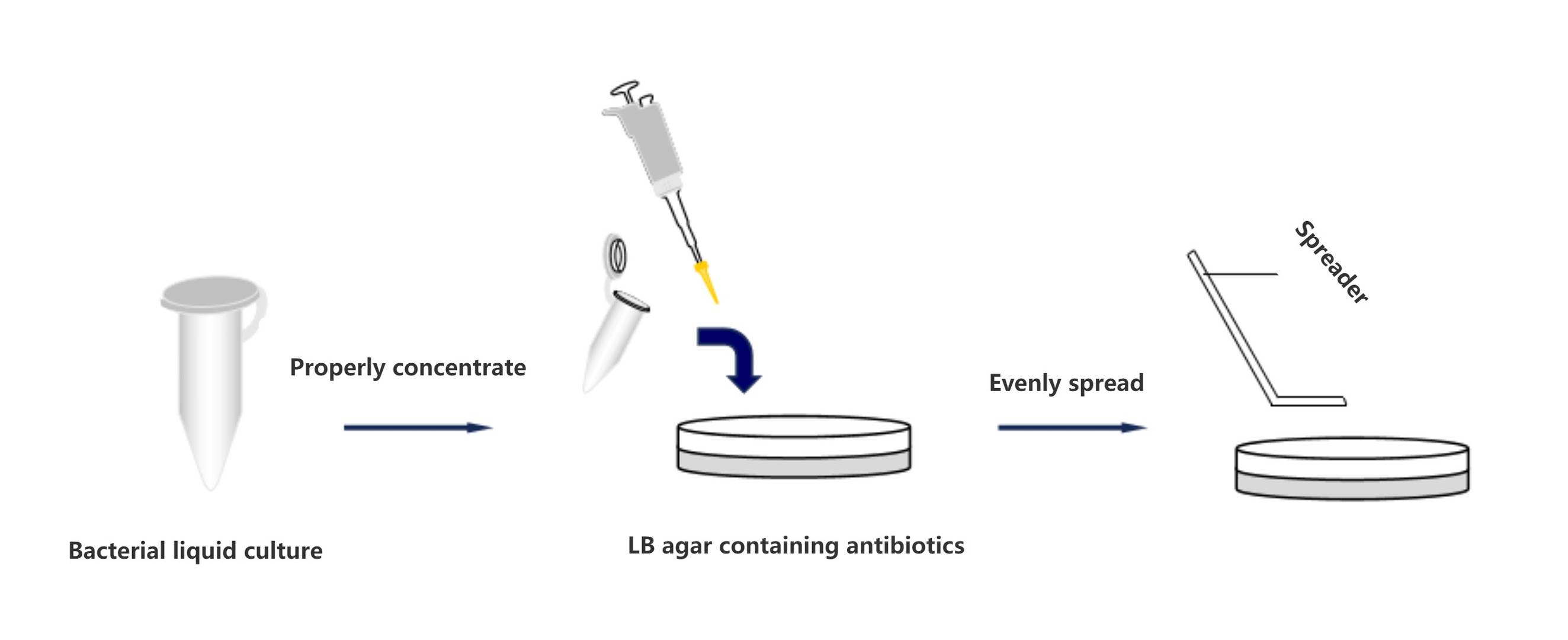
The incubation period for LB medium without antibiotics should be carefully regulated to prevent overgrowth of Gram-negative bacterial strains.
Uniform spreading and appropriate volume application are essential during plating to ensure the formation of an adequate number of individual bacterial colonies.
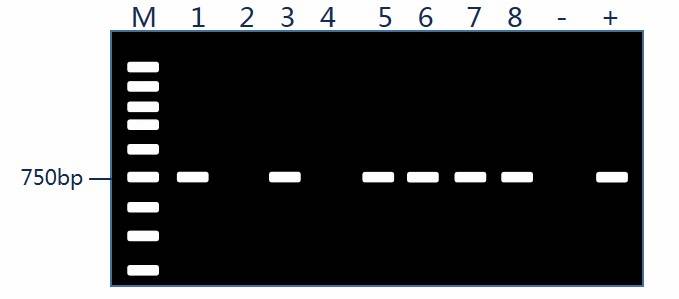
Screening of Recombinant Colonies
Upon formation of bacterial colonies, validation of their positivity is typically conducted using colony PCR methodology.
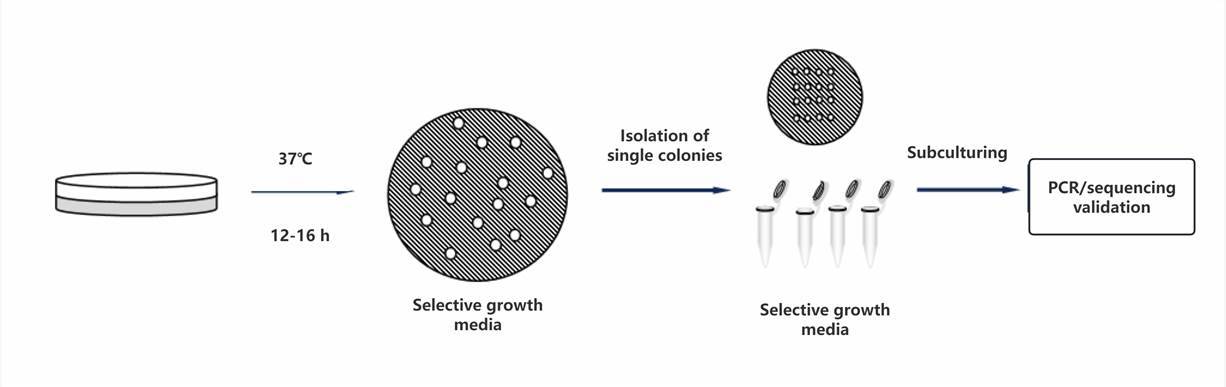
Different bacterial strains require varying incubation temperatures and durations to facilitate the formation of individual colonies as a guiding principle.
Positive recombinant colonies are preserved; therefore, it is customary to first conduct single colony expansion.

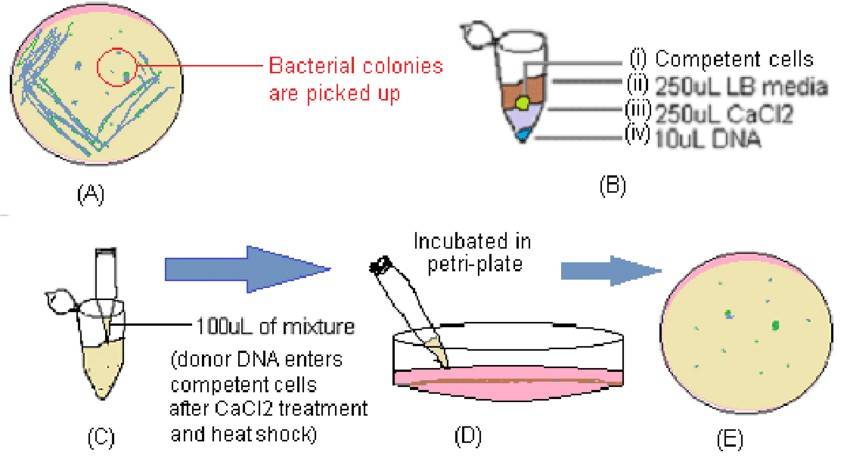 Illustration of main stages of bacterial transformation protocol (Ratul Banerjee 2022)
Illustration of main stages of bacterial transformation protocol (Ratul Banerjee 2022)