
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

RIP-Seq, or RNA Immunoprecipitation Sequencing, is an experimental method that integrates immunoprecipitation (IP) with high-throughput sequencing techniques to explore the interactions between RNA and proteins. In a RIP-Seq experiment, antibodies specific to a target protein are employed to precipitate RNA-protein complexes that bind to the target protein. This approach enables researchers to capture and isolate particular RNA molecules from complex biological samples, where these RNAs have direct physical associations with the target protein or protein complex. The precipitated RNA is extracted and reverse-transcribed into complementary DNA (cDNA), followed by deep analysis using high-throughput sequencing. This reveals interaction sites and potential functional implications of RNA-protein interactions.
RIP-Seq helps scientists see how RNA connects to proteins, giving insights into RNA functions after they are made. Consequently, it helps infer the roles these RNAs may play within the cell. This technique is extensively utilized in the study of RNA-binding proteins (RBPs) and in investigating interactions between various types of non-coding RNAs—such as long non-coding RNAs (lncRNAs) and microRNAs—and their associated binding proteins.
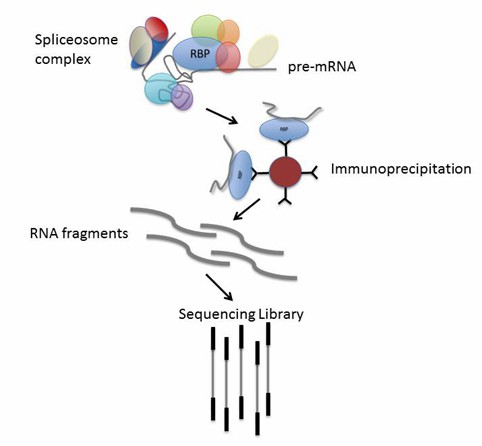 RNA immunoprecipitation (RIP-seq) done by targeting RNA binding proteins (RBPs). (Head, Steven R., et al. Biotechniques, 2014)
RNA immunoprecipitation (RIP-seq) done by targeting RNA binding proteins (RBPs). (Head, Steven R., et al. Biotechniques, 2014)
CD Genomics provides high-quality sequencing solutions tailored for epitranscriptomics and RNA-protein interaction studies. Our comprehensive services ensure precise and reliable results, supporting diverse research applications. Our services include:
Within the framework of RIP-Seq experiments, the preparation of the sequencing library is a critical determinant of data quality. The integrity and accuracy of the final sequencing data are inherently dependent on the caliber of the library preparation. The process includes extracting RNA, removing ribosomal RNA (rRNA), turning it into DNA, and then building a library for sequencing.
1. RNA Purity: The extraction and purification of RNA represent pivotal steps in the preparation of the library. Any contamination or degradation of RNA can significantly compromise the quality of subsequent sequencing data. Therefore, ensuring the integrity and purity of RNA within the immunoprecipitates is fundamental for the reliability of experimental outcomes. For details on sample preparation in RIP-Seq, refer to "RIP-Seq Sample Preparation Protocol."
2. Library Uniformity: Variations during library construction can introduce biases into the sequencing results. For instance, incomplete removal of rRNA may lead to inadequate enrichment and subsequent underrepresentation of target RNAs, thus affecting their identification and analysis.
3. Sequencing Depth Assurance: Attaining sufficient sequencing depth through high-throughput technologies is another critical objective in library preparation. Insufficient coverage can result in the omission of low-abundance RNAs. Hence, optimizing the library construction process to achieve uniformity and adequate coverage depth is vital for producing reliable and reproducible results.
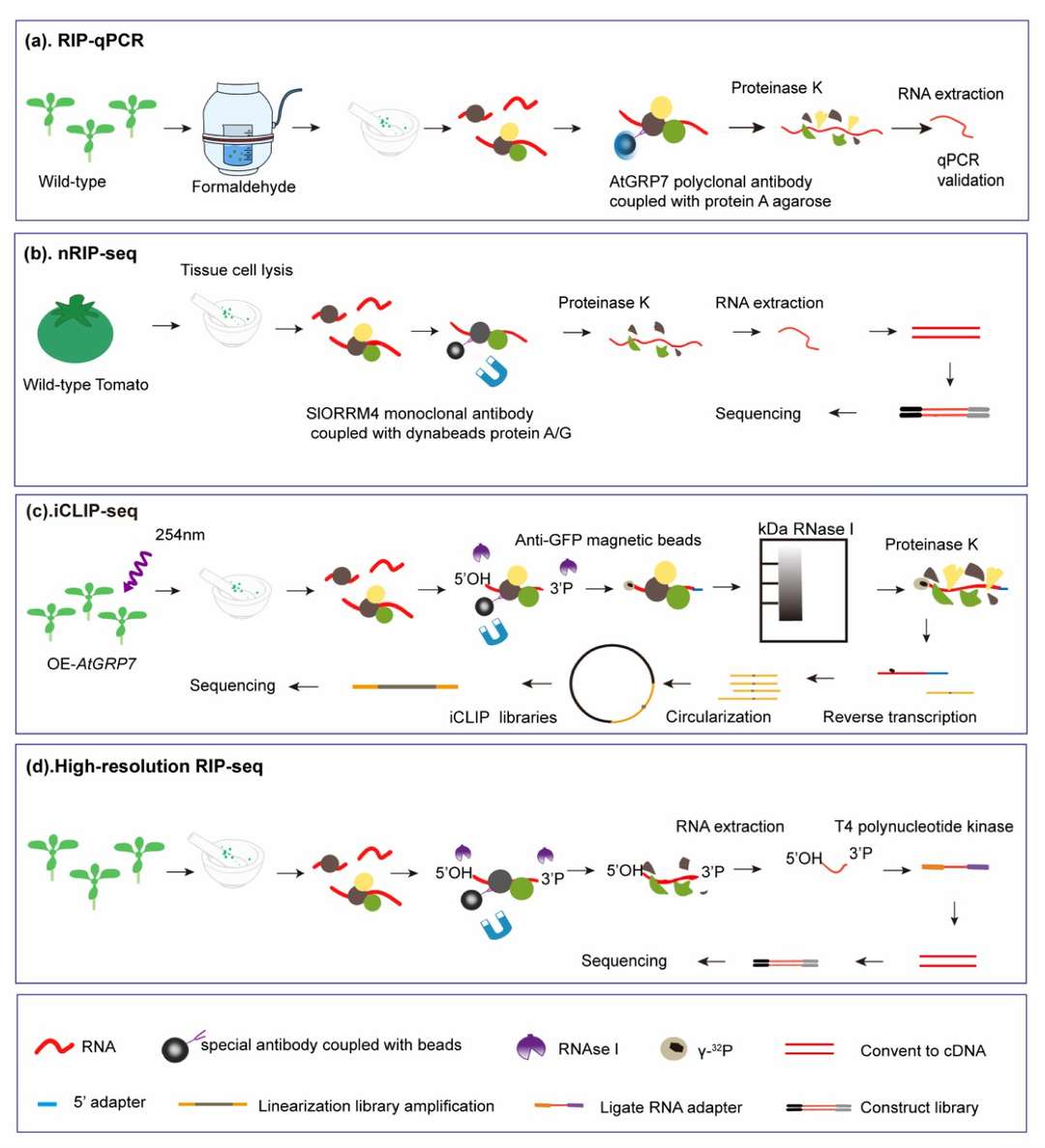 Workflow of RIP-qPCR, native RIP-seq, iCLIP and high-resolution RIP-seq analyses of plants. (Ma, Liqun, et al., International journal of molecular sciences, 2021)
Workflow of RIP-qPCR, native RIP-seq, iCLIP and high-resolution RIP-seq analyses of plants. (Ma, Liqun, et al., International journal of molecular sciences, 2021)
The preparation of a RIP-Seq library is a comprehensive, multi-faceted procedure that encompasses every stage from RNA extraction in cellular samples to high-throughput sequencing. Each step warrants meticulous attention to ensure the accuracy and quality of the resultant data.
Preliminary Preparations
Experimental Steps
Principal RIP/RIP-Seq Experimental Steps
1. Cell Collection and Crosslinking
2. Cell and Nuclear Lysis Consideration
3. Chromatin Shearing
4. RNA Immunoprecipitation
5. RNA Extraction and Analysis
Each step is explained clearly to show how complex preparing RIP-Seq libraries can be, essential for researchers aiming to investigate RNA-protein interactions with high fidelity.
The preparation of libraries for RNA Immunoprecipitation Sequencing (RIP-Seq) is crucial for effectively elucidating RNA-protein interactions. Three primary challenges in this process include RNA purification, antibody selection, and immunoprecipitation efficiency. Overcoming these challenges necessitates scientifically optimized strategies.
1. RNA Purification and Recovery
Challenge: The purity of RNA is fundamental to the quality of sequencing data. In RIP-Seq, the RNA is typically part of intricate protein assemblies, potentially complicating its recovery and purification.
Solution: Implement highly effective RNA extraction techniques, such as the Trizol method, to reliably isolate pure RNA from immunoprecipitated complexes. Moreover, the application of RNA stabilizers is recommended to prevent degradation and maintain RNA integrity throughout the procedure.
2. Antibody Selection
Challenge: The specificity and quality of antibodies are vital. Antibodies that fail to adequately bind to the target protein, or that bind non-specifically, can cause experimental failure.
Solution: Select high-quality, validated antibodies, preferably ChIP-grade, to ensure specificity and proper binding. When specific antibodies are unavailable, tagged antibodies may serve as an alternative, provided successful fusion of the tags with the target protein is achieved.
3. Efficiency of Immunoprecipitation
Challenge: The immunoprecipitation efficiency directly influences RNA capture. Inefficient procedures may result in inadequate enrichment of target RNA, thereby compromising sequencing precision.
Solution: Fine-tune the ratio of antibody to magnetic beads, and adjust experimental conditions like incubation time and temperature, to optimize the enrichment of RNA-protein complexes.
By addressing these challenges with targeted solutions, researchers can significantly enhance the reliability and accuracy of RIP-Seq, thus advancing the understanding of RNA-protein interactions and their roles in various biological processes.
Optimizing RIP-Seq for High-Quality Results
Achieving success in RIP-Seq experiments requires meticulous optimization at every stage, from experimental design to data analysis. Enhancing the specificity and efficiency of the assay through rigorous antibody and bead selection, fine-tuning experimental conditions, and leveraging advanced bioinformatics tools allows researchers to generate robust, biologically meaningful data.
Optimizing Antibody and Bead Selection
The choice of antibody is critical, as it must be highly specific to the target RNA-binding protein (RBP) to ensure reliable immunoprecipitation. Only thoroughly validated antibodies should be employed. Additionally, using high-efficiency magnetic beads optimized for protein-RNA complex binding enhances the capture and recovery of target RNA molecules, minimizing background noise.
Refining Experimental Conditions
Experimental conditions, including the composition of lysis buffers, incubation times, and temperatures, as well as antibody-to-bead ratios, must be precisely calibrated. Proper optimization ensures efficient immunoprecipitation while minimizing non-specific binding, thereby increasing the reproducibility and reliability of the results.
Advancing Data Analysis
Bioinformatics tools are indispensable for processing and interpreting RIP-Seq data. State-of-the-art algorithms can accurately map sequencing reads, identify RNA-protein interaction peaks, and annotate binding sites. These analyses uncover functional insights into RNA-protein interactions and reveal regulatory mechanisms at both gene-specific and transcriptome-wide scales.
RIP-Seq is a powerful technique for studying RNA-protein interactions, providing valuable insights into the intricate mechanisms of post-transcriptional regulation. By adhering to precise library preparation protocols and refining experimental workflows, this method can deliver high-quality data that advance our understanding of RNA-protein networks.
Looking forward, as sequencing technologies and bioinformatics platforms continue to evolve, RIP-Seq holds great promise for applications in single-cell studies and complex biological systems. These advancements will enable deeper exploration of RNA regulatory networks and their roles in health and disease. Through ongoing optimization of experimental and analytical strategies, RIP-Seq will remain a vital tool for elucidating RNA-protein interactions, disease mechanisms, and gene expression regulation.
The most prominent techniques for studying RNA-RBP interactions are RNA Immunoprecipitation Sequencing (RIP-Seq) and Crosslinking Immunoprecipitation Sequencing (CLIP-Seq). For a detailed comparison of these methods, refer to the article: RIP-Seq vs. CLIP-Seq: Introduction, Advantages, and Applications.
References:


