Viruses, acting as both symbionts and pathogens, contribute to tumorigenesis through mechanisms such as genomic integration, immune modulation, and inflammation induction. Classical oncogenic viruses drive malignancy by integrating into the host genome, suppressing tumor suppressor pathways, or activating pro-oncogenic signals. Viral DNA has been specifically identified in tumor tissues of colorectal cancer (CRC), gastric cancer, and other malignancies. Recent studies highlight the role of the gut virome in CRC progression via direct infection, microbiota regulation, or immune interactions, where phage dysbiosis triggers bacterial lysis and subsequent DNA damage. The long-term co-evolution of viruses and hosts has fostered unique immune-balancing strategies. Targeting viral immune evasion mechanisms-such as using DNA methyltransferase inhibitors like decitabine to induce viral antigen expression and activate interferon responses through TLRs/MDA5 pathways-can enhance antitumor immunotherapy. Virome research not only deepens our understanding of tumorigenesis but also pioneers innovative diagnostic and therapeutic strategies for cancer.
Molecular Mechanisms of Viral Carcinogenesis
Viruses, existing at the crossroads of symbiosis and pathogenicity, have emerged as master manipulators of cellular machinery, driving carcinogenesis through multifaceted mechanisms. Their ability to directly damage DNA, subvert immune surveillance, and induce chronic inflammation positions them as potent oncogenic agents [1]. Globally, the World Health Organization (WHO) estimates that 15% of cancers-including cervical, hepatic, and gastric malignancies-are causally linked to persistent viral infections. Among these, Epstein-Barr virus (EBV), human papillomavirus (HPV), and hepatitis B virus (HBV) stand out as archetypal oncoviruses, each employing distinct strategies to hijack eukaryotic cellular processes.
Notably, viral DNA exhibits striking tissue specificity in tumor environments. For instance, HPV, polyomaviruses, and herpesviruses are enriched in colorectal cancer (CRC) tissues but absent in adjacent normal mucosa, while gastric tumors harbor unique viral signatures [2]. This spatial restriction suggests a finely tuned evolutionary adaptation, where viruses exploit tissue-specific vulnerabilities to establish oncogenic niches.
The genomic footprint of viruses in cancer is further illuminated by integration studies. Pioneering work by X. Chen et al. revealed widespread viral sequence integration across diverse malignancies, including hepatocellular carcinoma and non-Hodgkin lymphoma [3]. Such integration events frequently cluster near genomic fragile sites-regions prone to breakage during replication-amplifying chromosomal instability.
Service you may interested in
Resource
Genomic Integration and Tumor Suppressor Disruption
Viral integration into tumor suppressor loci represents a "genetic sabotage" strategy. HPV, for example, preferentially integrates into TP53 (chromosome 17p13) and RB1 (13q14), truncating these guardians of genomic integrity (Figure 1). This inactivation unleashes cell cycle progression and apoptotic resistance, hallmarks of malignancy. Recent single-cell sequencing studies reveal that >60% of HPV+ cervical cancers harbor such disruptive integrations, correlating with poor prognosis [4].
Subversion of Tumor Suppressor Pathways
Viral proteins often mimic host signaling molecules to dysregulate critical pathways. EBV's latent membrane protein 1 (LMP1) constitutively activates NF-κB, upregulating anti-apoptotic Bcl-2 family proteins while suppressing pro-death signals like BIM. Similarly, HBV's HBx protein stabilizes β-catenin, driving Wnt pathway hyperactivation and hepatic stem cell expansion.
Oncogenic Signal Amplification
Chronic inflammation induced by viral persistence creates a mutagenic microenvironment. HBV-associated hepatitis fosters oxidative stress through cytochrome P450 activation, generating DNA-damaging ROS. Concurrently, viral non-structural proteins (e.g., HCV's NS5A) directly interact with PI3K/Akt/mTOR axes, fueling metabolic reprogramming in pre-neoplastic lesions.
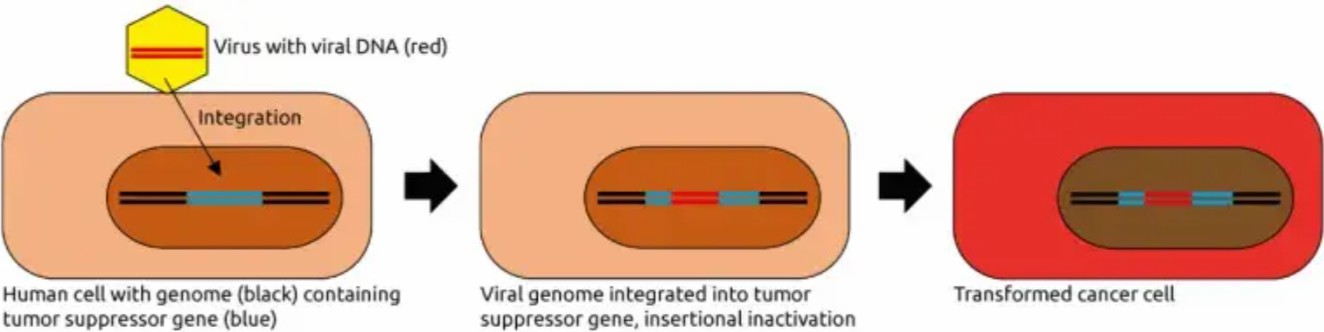 Figure 1:Mechanisms of eukaryotic virus-induced carcinogenesis. [4].
Figure 1:Mechanisms of eukaryotic virus-induced carcinogenesis. [4].
These insights underscore the dual utility of viral genomics: as a diagnostic tool for identifying occult infections and as a therapeutic roadmap for precision oncology.
Virome-Microbiome Interactions and Tumor Immunotherapy
Gut Virome Ecology and Tumorigenesis
The gut virome-a dynamic consortium of viral particles-has emerged as a critical modulator of carcinogenesis. While bacteriophages traditionally have been bacterial predators, their role in shaping microbial ecology carries profound oncogenic implications. In CRC, phage dysbiosis disrupts the delicate balance between lytic and temperate phage populations. As depicted in Figure 2, rampant lytic phage activity decimates keystone bacterial species (e.g., Faecalibacterium prausnitzii), enabling pathobiont overgrowth.
The collateral damage extends beyond microbial shifts: phage-induced bacterial lysis releases endotoxins (e.g., LPS) and genotoxic metabolites. E. coli strains carrying pks islands secrete colibactin-a microbial toxin that alkylates DNA, inducing crosslinks and replication errors. Strikingly, colibactin-associated mutational signatures (e.g., tandem duplications at AA/TT dinucleotides) are detected in 67% of CRC genomes, directly linking phage activity to genomic catastrophe [5].
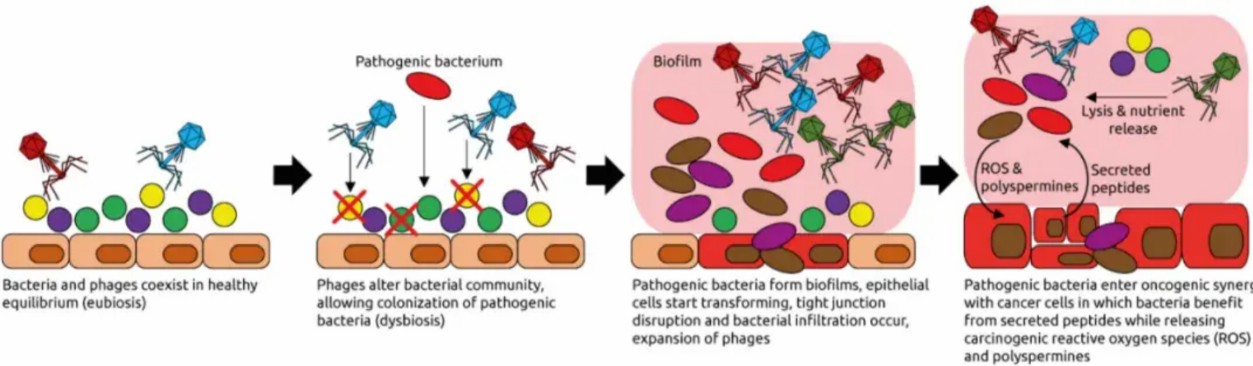 Figure 2: Phage-induced tumorigenesis. Dysbiotic phages lyse commensal bacteria, enabling pathobiont biofilm formation. Biofilm-derived ROS and colibactin drive epithelial DNA damage [5].
Figure 2: Phage-induced tumorigenesis. Dysbiotic phages lyse commensal bacteria, enabling pathobiont biofilm formation. Biofilm-derived ROS and colibactin drive epithelial DNA damage [5].
Bidirectional Host-Virome Interactions
The virome's carcinogenic potential is amplified through bidirectional crosstalk with host systems (Figure 3). In IBD-associated CRC, enteric viruses like norovirus exploit compromised mucosal barriers, triggering a feedforward cycle of inflammation. Viral RNA sensors (RIG-I/MDA5) activate NLRP3 inflammasomes, releasing IL-1β and IL-18-cytokines that simultaneously promote epithelial proliferation and suppress anti-tumor T cells.
Conversely, phages indirectly modulate host physiology by reprogramming bacterial metabolism. For instance, Clostridium phages enhance bacterial conversion of dietary choline to trimethylamine (TMA), which hepatocytes oxidize to pro-atherogenic TMAO. Elevated TMAO levels correlate with hepatic CYP7A1 suppression, impairing bile acid detoxification and fostering DNA adduct formation-a known driver of cholangiocarcinoma [6].
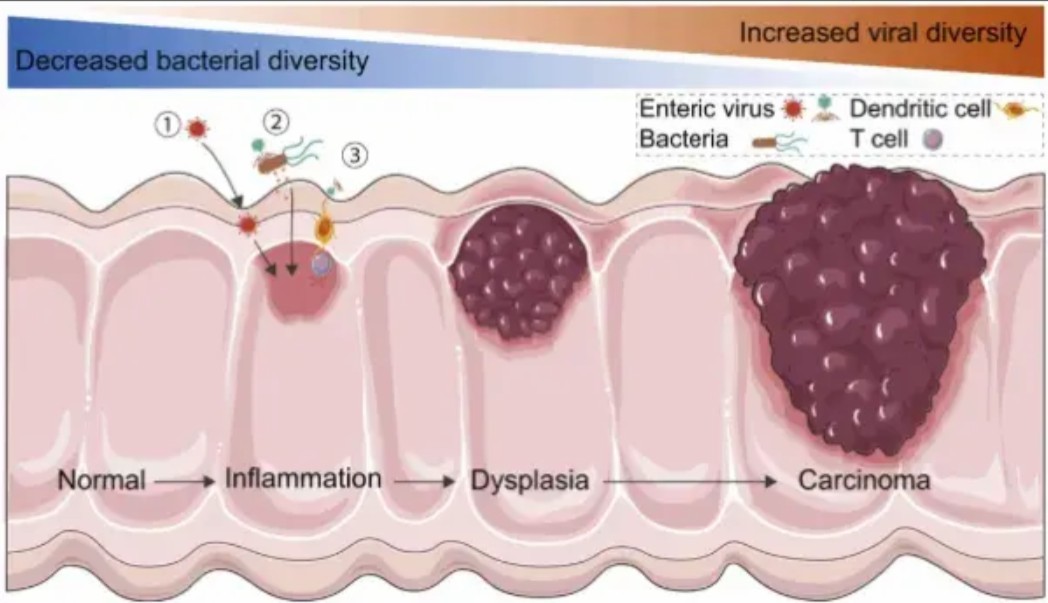 Figure 3: Host-virome interactions in intestinal inflammation and cancer. [5].
Figure 3: Host-virome interactions in intestinal inflammation and cancer. [5].
The therapeutic potential of virome modulation is exemplified by fecal microbiota transplantation (FMT). In a landmark trial, FMT from PD-1 inhibitor responders restored Bacteroides-phage networks in refractory melanoma patients, increasing CD8+ TIL infiltration and doubling response rates [7]. This highlights the virome's role as a tunable "immunomodulatory switch."
Synergy Between Antiviral Immunity and Tumor Immunotherapy
The co-evolutionary arms race between viruses and hosts has yielded unexpected therapeutic opportunities. Latent viral reactivation strategies exploit the immune system's exquisite sensitivity to viral components:
Human Endogenous Retroviruses (HERVs): Once considered genomic "junk," HERVs comprise 8% of human DNA. DNA methyltransferase inhibitors (e.g., decitabine) demethylate HERV long terminal repeats (LTRs), unleashing dsRNA transcripts. These viral mimics activate TLR3/7 and MDA5, triggering a cascade of type I IFN production, dendritic cell maturation, and tumor-specific CD8+ T-cell expansion (Figure 4). In phase I trials, decitabine combined with anti-PD-1 increased objective responses in HERV-K+ ovarian cancers by 40% compared to monotherapy [4].
Oncolytic Viral Synergy: Seasonal influenza vaccination primes dendritic cells via TLR7/MyD88 signaling, enhancing cross-presentation of tumor antigens. In EBV+ nasopharyngeal carcinoma, this "bystander activation" amplified tumor-infiltrating lymphocyte (TIL) clonality, correlating with prolonged progression-free survival [7].
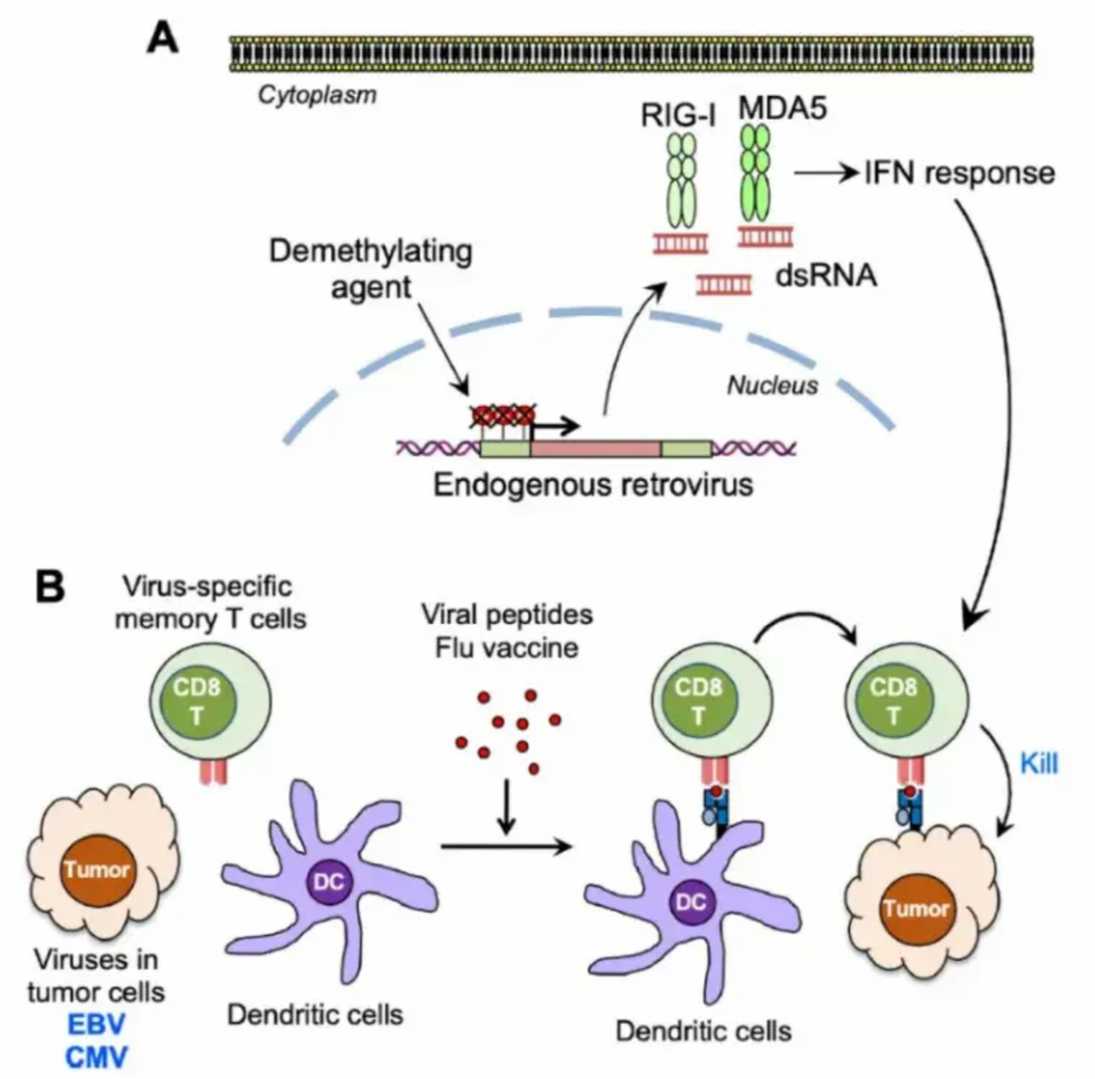 Figure 4: Antiviral immune activation enhances antitumor immunotherapy. [7].
Figure 4: Antiviral immune activation enhances antitumor immunotherapy. [7].
This paradigm of virome-directed immunotherapy redefines viruses from foes to allies in the oncological armamentarium.
Conclusions and Future Perspectives
Virome research has irrevocably altered our understanding of carcinogenesis, positioning viral communities as both biomarkers and therapeutic targets. Yet, critical frontiers demand attention:
Technological Innovation:
oThird-Generation Sequencing: Nanopore and PacBio platforms enable full-length viral genome assembly, resolving integration sites with single-base precision.
oCRISPR-Based Virome Editing: Engineered phages carrying Cas9 could selectively eradicate oncobiotic bacteria (e.g., Fusobacterium nucleatum) while preserving commensals.
Functional Viromics:
oHigh-Throughput Screens: ORFeome libraries of viral genes could identify novel oncoproteins (e.g., HBV's HBx interactome).
oSpatiotemporal Mapping: Multiplexed imaging (CODEX/IMC) will visualize viral replication niches within tumor ecosystems.
Translational Integration:
oVirome Biomarkers: Plasma virome signatures (e.g., Anellovirus load) may predict immunotherapy response.
oPhage Cocktails: Clinically approved phage preparations (e.g., PYOPAX) could prevent chemotherapy-induced dysbiosis.
Emerging tools like tumor virome organoids and synthetic viral vectors promise to bridge bench-to-bedside gaps. As spatial transcriptomics unravels the "geography" of viral activity, and AI-driven platforms (e.g., VirScan) decode host-virome interactomes, we approach an era of precision ecological oncology-where modulating microbial networks becomes as routine as genomic profiling.
References:
- Read, S. A., & Douglas, M. W. (2014). Virus induced inflammation and cancer development. Cancer letters, 345(2), 174–181. https://doi.org/10.1016/j.canlet.2013.07.030
- Coker O. O. (2022). Non-bacteria microbiome (virus, fungi, and archaea) in gastrointestinal cancer. Journal of gastroenterology and hepatology, 37(2), 256–262. https://doi.org/10.1111/jgh.15738
- Chen, X., Kost, J., Sulovari, A.. (2019). A virome-wide clonal integration analysis platform for discovering cancer viral etiology. Genome research, 29(5), 819–830. https://doi.org/10.1101/gr.242529.118
- Broecker, F., & Moelling, K. (2021). The Roles of the Virome in Cancer. Microorganisms, 9(12), 2538. https://doi.org/10.3390/microorganisms9122538
- Wang, Z., Guo, K., Liu, Y., Huang, C., & Wu, M. (2022). Dynamic impact of virome on colitis and colorectal cancer: Immunity, inflammation, prevention and treatment. Seminars in cancer biology, 86(Pt 2), 943–954. https://doi.org/10.1016/j.semcancer.2021.10.004
- Cullin, N., Azevedo Antunes, C., Straussman, R.. (2021). Microbiome and cancer. Cancer cell, 39(10), 1317–1341. https://doi.org/10.1016/j.ccell.2021.08.006
- Pyeon, D., Vu, L., Giacobbi, N. S., & Westrich, J. A. (2020). The antiviral immune forces awaken in the cancer wars. PLoS pathogens, 16(9), e1008814. https://doi.org/10.1371/journal.ppat.1008814

 Sample Submission Guidelines
Sample Submission Guidelines


