Within the realm of global health challenges, viral pathogens from the influenza family pose substantial risks through their capacity to trigger extensive outbreaks and recurring seasonal disease patterns. The development of successful strategies to combat and limit viral transmission depends fundamentally on comprehensive knowledge regarding influenza strain variations and their underlying genetic composition.
What is Influenza Virus
Respiratory infections caused by influenza viruses primarily affect the upper airways and respiratory tract, with occasional lung involvement. These pathogens can trigger health effects that vary significantly in severity. As members of Orthomyxoviridae, these viruses possess distinctive RNA genetic material that is both segmented and negative-sense in nature.
The genomic structure of influenza comprises distinct RNA components, with each segment coding for specific viral components. When different viral strains simultaneously occupy one host cell, their segmented genetic material enables recombination, potentially generating novel variants. Research into genomic characteristics and viral recombination processes plays a crucial role in surveillance of viral changes, identification of emerging pandemic candidates, and advancement of preventive and therapeutic interventions.
Service you may interested in
Resources
Types of Influenza Viruses
Influenza viruses are categorized into four main types: A, B, C, and D. Each type exhibits distinct characteristics regarding subtypes or lineages, genetic behavior, and impact on human and animal health.
Influenza A Virus
Subtypes and Their Characteristics
Influenza A viruses are classified into subtypes based on two surface proteins: hemagglutinin (H) and neuraminidase (N). There are 18 known hemagglutinin subtypes and 11 neuraminidase subtypes, leading to various HN combinations, such as H1N1 and H3N2. These subtypes are identified in both human and animal populations, with wild aquatic birds serving as the primary natural reservoir(Allen, J. D.,et.al,2018).
Role in Pandemics
Influenza A viruses are notorious for causing pandemics. Their segmented RNA genome allows for genetic reassortment, especially when different subtypes infect the same host. This reassortment can result in new viral strains with pandemic potential. Historical pandemics, such as the 1918 H1N1 "Spanish flu" and the 2009 H1N1 "swine flu," were caused by novel reassortant influenza A viruses.
Genetic Reassortment
Genetic reassortment in influenza A occurs when genome segments from distinct strains mix within a co-infected host cell. This process can lead to the emergence of new viral strains with unique properties, influencing viral evolution and posing challenges for public health.
Influenza B Virus
Lineages and Clades
Influenza B viruses are divided into two primary lineages: B/Victoria and B/Yamagata. Each lineage is further categorized into various clades based on genetic and antigenic properties. These lineages have distinct evolutionary patterns and antigenic drift rates.
Genetic Stability Compared to Influenza A
Influenza B viruses exhibit greater genetic stability than influenza A viruses. The B/Yamagata lineage, for instance, has an average antigenic drift rate of 6.3 to 7.2 years, while the B/Victoria lineage shows faster drift rates, averaging 3.9 to 5.1 years. In contrast, influenza A viruses undergo more rapid antigenic changes, contributing to their higher pandemic potential.
Impact on Human Health
While influenza B viruses primarily infect humans and contribute to seasonal flu epidemics, they are generally associated with less severe disease compared to influenza A viruses. Notably, influenza B viruses have not been linked to pandemics, partly due to their limited host range and lower propensity for genetic reassortment.
Influenza C Virus
Mild Illness and Limited Epidemic Potential
Influenza C viruses cause mild respiratory illnesses and are not known to cause epidemics. Infections are typically sporadic, and the virus has a limited impact on public health.
Genetic Structure
Influenza C viruses have a distinct genetic structure compared to influenza A and B viruses. They possess seven RNA segments, encoding nine proteins, and utilize a different surface glycoprotein called hemagglutinin-esterase-fusion (HEF) protein, which combines the functions of hemagglutinin and neuraminidase found in other influenza types.
Influenza D Virus
Primary Impact on Cattle
Influenza D viruses primarily affect cattle and are not known to infect or cause illness in humans. They are the least studied among the influenza viruses and currently pose no known threat to human health.
Limited Human Infection
To date, there is no evidence of influenza D virus causing infections in humans. Research continues to monitor this virus for any potential zoonotic transmission, but it currently remains an animal health concern without implications for human health.
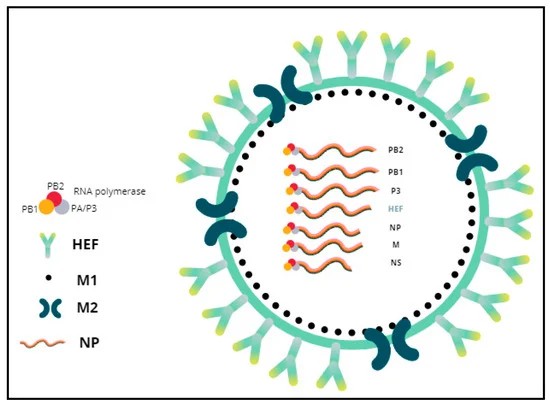 Figure1. General morphology of influenza D virus. (Ruiz, M.;et.al,2022)
Figure1. General morphology of influenza D virus. (Ruiz, M.;et.al,2022)
Understanding the distinct characteristics of each influenza virus type is crucial for developing targeted prevention and treatment strategies, as well as for anticipating and mitigating potential public health impacts.
Genetic Structure of Influenza Viruses
Influenza viruses possess unique genetic structures that contribute to their ability to cause widespread illness and adapt over time. Understanding their genomic composition, key proteins, and mechanisms of genetic variation is essential for developing effective prevention and treatment strategies.
Summary of the Influenza Virus Genome
Single-Stranded RNA Segments: The influenza virus genome consists of multiple segments of single-stranded RNA (ssRNA). For instance, influenza A and B viruses have eight RNA segments, while influenza C viruses have seven. Each segment encodes one or more proteins essential for the virus's life cycle.
Negative-Sense RNA: These RNA segments are of negative-sense orientation, meaning they are complementary to the viral mRNA and must be transcribed into positive-sense mRNA by the viral RNA-dependent RNA polymerase before translation into proteins. This negative-sense nature necessitates the packaging of the viral polymerase within the virion to initiate replication upon infection.
Key Proteins and Their Functions
Hemagglutinin (HA)
Hemagglutinin is a surface glycoprotein responsible for binding the virus to sialic acid receptors on the host cell surface, facilitating viral entry. It also mediates the fusion of the viral envelope with the host cell membrane, a critical step for the release of the viral genome into the host cell.
Neuraminidase (NA)
Neuraminidase is another surface enzyme that facilitates the release of newly formed virions from the host cell by cleaving sialic acid residues. This activity prevents the aggregation of virions and promotes the spread of the virus within the host.
Other Structural and Nonstructural Proteins
Influenza viruses encode several other proteins with diverse functions:
Nucleoprotein (NP): Encapsidates the viral RNA, forming ribonucleoprotein complexes essential for genome replication and transcription.
Polymerase Complex (PB1, PB2, PA): These subunits constitute the viral RNA-dependent RNA polymerase responsible for replicating and transcribing the viral genome. The PA subunit, for example, has endonuclease activity crucial for viral mRNA synthesis.
Matrix Proteins (M1 and M2): M1 is involved in virion assembly and stability, while M2 functions as an ion channel important for uncoating the virus during entry.
Nonstructural Proteins (NS1 and NS2): NS1 modulates host immune responses and enhances viral replication, whereas NS2 (also known as NEP) is involved in the nuclear export of viral ribonucleoproteins.
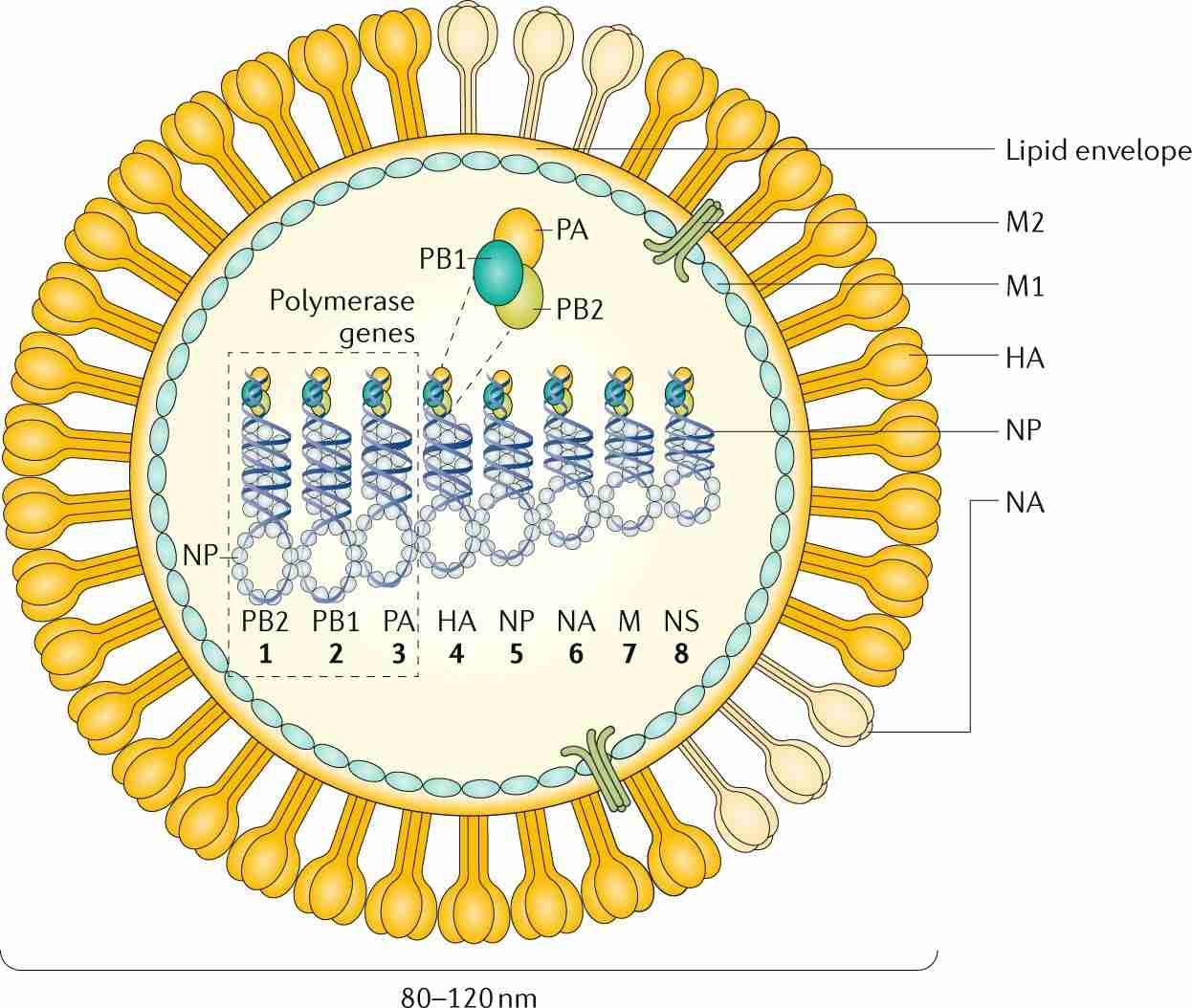 Figure 2.The structure of Influenza A and influenza B.(Krammer, F., et.al,2018)
Figure 2.The structure of Influenza A and influenza B.(Krammer, F., et.al,2018)
Genetic Diversity and Mutation Rates
Antigenic Drift
Antigenic drift refers to the gradual accumulation of mutations in the viral genome, particularly in the HA and NA genes. These mutations can alter antigenic sites, enabling the virus to evade host immune responses and necessitating periodic updates to influenza vaccines.
Antigenic Shift
Antigenic shift is a process unique to influenza A viruses, involving the reassortment of gene segments when two different strains infect the same host cell. This can result in the emergence of a novel subtype with a significantly different antigenic profile, potentially leading to pandemics due to a lack of population immunity.
High Mutation Rates and Evolutionary Dynamics
Influenza viruses exhibit high mutation rates due to the lack of proofreading mechanisms in their RNA-dependent RNA polymerase. This rapid evolution allows them to adapt quickly to selective pressures, such as host immune responses and antiviral treatments, contributing to their persistence and variability.
Understanding the genetic structure and variability of influenza viruses is crucial for monitoring their evolution, developing effective vaccines, and implementing appropriate public health interventions.
Genome Sequencing and Genetic Characterization
Genome sequencing and genetic characterization are pivotal in understanding influenza viruses, enabling effective monitoring, prevention, and treatment strategies.
Methods of Genome Sequencing
Sanger Sequencing
Sanger sequencing, developed in the 1970s, was the first method used for partial influenza A virus genome sequencing. It employs chain-terminating nucleotides to determine DNA sequences and has been fundamental in early influenza research. However, its limitations in throughput and scalability have led to the adoption of more advanced techniques.
Next-Generation Sequencing (NGS)
NGS technologies have revolutionized influenza virus research by allowing high-throughput, comprehensive genome analysis. Methods such as amplicon sequencing enable efficient whole-genome sequencing of influenza A and B viruses. NGS facilitates deep sequencing to characterize virus populations, identify viral variants, and monitor genetic diversity.
Importance of Genetic Characterization
Monitoring Viral Evolution
Genetic characterization provides insights into the evolution of influenza viruses, including mutation rates and patterns. High-throughput sequencing data allow for detailed analysis of genetic diversity, which is crucial for understanding viral adaptation and preparing for future outbreaks.
Assessing Vaccine Efficacy
By analyzing genetic changes in circulating influenza strains, researchers can assess how well current vaccines match these strains. This information guides the selection of vaccine components, ensuring optimal protection against prevalent virus variants.
CDC
Identifying Drug Resistance
Genetic sequencing can detect mutations associated with resistance to antiviral drugs. Early identification of such mutations enables timely adjustments to treatment protocols and the development of new therapeutic strategies.
Conclusion
In conclusion, the genetic diversity of influenza viruses, driven by their segmented RNA genomes and high mutation rates, poses a significant challenge to global health. Influenza A viruses, with their potential for genetic reassortment and pandemics, are the most concerning, while influenza B viruses contribute to seasonal epidemics. Influenza C and D viruses, though less impactful, still require monitoring. Advances in genome sequencing have enhanced our ability to track viral evolution, assess vaccine efficacy, and identify drug resistance. Continued research into influenza's genetic makeup and evolution is essential for developing effective prevention and treatment strategies, ensuring preparedness for future outbreaks and safeguarding public health.
References:
- Allen, J. D., & Ross, T. M. (2018). H3N2 influenza viruses in humans: Viral mechanisms, evolution, and evaluation. Human vaccines & immunotherapeutics, 14(8), 1840–1847. https://doi.org/10.1080/21645515.2018.1462639
- Ruiz, M.; Puig, A.; Bassols, M.; Fraile, L.; Armengol, R. Influenza D Virus: A Review and Update of Its Role in Bovine Respiratory Syndrome. Viruses 2022, 14, 2717. https://doi.org/10.3390/v14122717
- Krammer, F., Smith, G.J.D., Fouchier, R.A.M. et al. Influenza. Nat Rev Dis Primers 4, 3 (2018). https://doi.org/10.1038/s41572-018-0002-y

 Sample Submission Guidelines
Sample Submission Guidelines


