The advent of metagenome-assembled genomes (MAGs) has transformed microbial genomics, as it allows the specific reconstruction of individual genomes from metagenomic data. MAGs give access to uncultivated microbial organisms that can be potential resources, highlighting that this practice is able to tap into an infinite array of unassigned life forms that cannot be cultivated by traditional culture methods. This approach has allowed complex ecosystems like soil, oceans and the human microbiome - where thousands of species live and interact - to be studied.
MAGs are generated from metagenomic datasets by assembling available short or long reads using commonly used technologies, enabling high-throughput sequencing, and organizing contigs into genome-level units using computational approaches. These genomes are extremely useful for deciphering the metabolic potential, ecological function and evolutionary relationships of microbes in managed and natural ecosystems. And as sequencing becomes increasingly affordable and accessible, MAGs are proving to be an essential tool to deciphering microbial life. These serve as the basis for separating complicated microbial interactions, identifying novel pathways, and exploring the functional capacities of microbial communities. This revolutionary power to examine genomes without cultivation processes has far-reaching consequences for environmental biology, human health, and industrial uses.
The additional MAGs help to solve the "microbial dark matter" problem as they reconstruct genomes from species of organisms that have not been cultivated in the laboratory. These discoveries have also elucidated ecological and evolutionary processes, highlighting novel microbial lineages and metabolic pathways. Resolving genomes from mixed microbial communities has also allowed researchers to learn more about symbiotic relationships and competition between microorganisms. Here, we presented an integrated review on MAGs in order to provided a brief introduction for the understanding and application of MAGs.
Service you may interested in
Resource
What is MAG Reconstruction
MAG reconstruction refers to the assembly of metagenomic data into genome fragments. Yet, due to the complexity and heterogeneity of microbial communities, this process is further complicated.
- Metagenomic Sequencing: In metagenomic sequencing, the entire sequence of genetic material from a sample is sequenced. This method characterizes the genomic diversity of complete microbial communities, encompassing bacteria, archaea, viruses and eukaryotes. Because metagenomics does not isolate individual organisms, it reflects a more holistic view of the genetic landscape of the ecosystem.
- Assembly and Binning: Metagenomic assembly refers to building up contiguous sequences (contigs) from raw sequencing reads. These contigs are subsequently allocated into sequence- and coverage-based bins that reflect draft genomes of individual species. In the case of mixed shotgun metagenome sequencing data, binning is vital for extracting genetic material from the individual organisms that participated in the experiment, particularly for hyper-diverse microbial environments.
- Quality Assessment: These metrics include completeness, contamination and genome fragmentation to assess the quality of MAGs. Good quality MAGs are at least 90% complete with less than 5% contamination, making them useful for functional analyses and comparative studies. These metrics are evaluated by software such as CheckM and BUSCO which perform indexing of single-copy ortholog recovery and genomic contamination levels
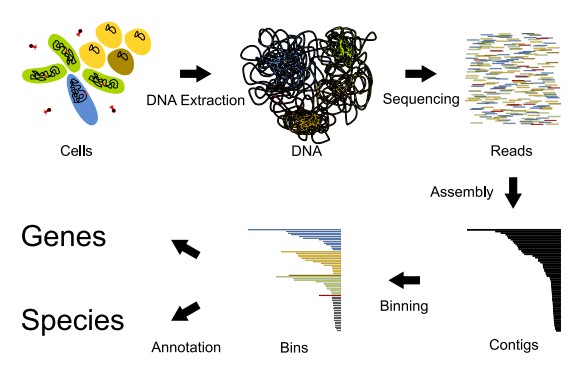 Metagenome analysis scheme (Goussarov, G. et al 2022).
Metagenome analysis scheme (Goussarov, G. et al 2022).
Current Challenges
- Community Complexity: There are thousands of species with different abundances in a microbial community, making assembly and binning challenging. Genome recovery is biased towards high-abundance species, leading to low-abundance organisms being underrepresented in the data. This puts a demand on the careful optimization of the sampling strategies and sequencing depth.
- Repetitive Elements: Repeated elements within and between genomes complicate the appropriate contig assembly. Transposable elements or conserved operons, for example, introduce ambiguities in read mapping and contig linking. Algorithms such as Flye and Canu that combine long-read sequencing data have worked well to resolve these regions.
- Contamination: The presence of sequencing artifacts and overlapping signal from closely related species can introduce contamination in highly similar MAGs. This further impairs the functional annotations and downstream analyses. These bins can be refined using additional tools such as MetaWRAP which further remove contaminants and refine the genome bins.
- Computational Requirements: The vast dimensional nature of metagenomic datasets necessitates high-dimensional computational resources for assembly, binning, and refinement. Datasets with billions of reads which need high memory systems and parallel processes to get juggled. The increasingly prevalent use of cloud computing solutions and optimized pipelines is making it possible to scale MAG reconstruction.
Technologies and Computational Methodologies
The success of MAG reconstruction has thus been driven by ever-evolving hardware meta- and algorithmic innovations. With specific sequencing platforms and data analysis software, all these elements contribute to the generation of high-quality MAGs.
Sequencing Platforms
Emerging tools have provided strategy to address the issues in MAGs. For example, Illumina sequencing have been applied in metagenomic studies on accounts of its lower cost and scalability today. However, its short reads tend to be unable to perform well in highly repetitive regions.
- Long-read Sequencing (PacBio and Nanopore): Produce reads that can stretch out over tens of kilobases, critical for covering over repetitive regions to better assemble complex genomes. PacBio HiFi reads, for example, provide the long read lengths while giving high accuracy, allowing for improvements in both continuity and correctness in assemblies.
- Hybrid Approaches: Merging short and long reads maximizes the accuracy and continuity of assemblies, especially in complex microbial communities. Hybrid approaches take advantage of the error-correcting abilities of short reads and the structural information offered by long reads. MaSuRCA and SPAdes are commonly utilized tools for this.
Assembly Tools
- MEGAHIT and MetaSPAdes: Tailored towards metagenomic data; do a pretty good job at short-read datasets. MEGAHIT was designed for ultra-large data sets, while MetaSPAdes provides error correction and an iterative graph construction framework for metagenomic assembly.
- Flye and Canu: Specialized long-read assembly tools that can be useful in low-complexity metagenomes to construct contiguous sequences. Flye can deal with noisy long reads, so handles Nanopore data, while Canu deals with PacBio assemblies.
Binning Algorithms
- Composition-based Methods: MetaBAT and CONCOCT are examples of composition-based tools that cluster contigs based on metrics of sequence composition, including GC content and tetranucleotide frequencies. These methods perform well at distinguishing genomes in communities of intermediate diversity.
- Coverage-based Methods: Tools like MaxBin exploit the differential coverage in each sample to enhance binning accuracy. By looking at coverage differences these tools can distinguish genomes with identical sequence compositions.
- Hybrid Methods: Some algorithms such as DAS Tool combine different binning methods in order to optimise and improve the quality of genome bins. This combination provides higher recall and accuracy on genome bins identification, in highly diverse datasets.
Refinement Tools
- CheckM: Assess completeness and contamination to assess MAG quality. It is valuable for monitoring the utility of MAGs in downstream analyses.
- Assembly tools: MetaWRAP and Anvi'o have workflows for reassembly and refinement of MAGs to improve accuracy and usability. Based on this, these tools allow for the visualization and in-depth exploration of metagenomic data, allowing for improved interpretation of the results.
Application for Metagenome-Assembled Genomes
From environmental science to medicine, MAGs have transformative applications across multiple disciplines. They are the key to reveal both uncultured microbial diversity and to clarify microbial functions, and they play a role as an aperture to the future evolutionary path of our planet.
Environmental Microbiology
MAGs allow for the investigation of microbial assemblages across various environments including soil, marine and extreme habitats. Researchers woven genomes from the data and mapped metabolic pathways that harness nutrient cycling, carbon sequestration, and pollutant degradation. Thus, MAGs from environmental samples like deep-sea sediments can provide new insights into the genetic basis of biogeochemical processes such as methane metabolism and hydrocarbon degradation. Such insights can inform the biogeochemical cycles and biotechnological processes.
Human Microbiome Studies
In human health, MAGs offer information about the structure and function of the human microbiome. They have played a crucial role in discovering microbial species linked to disorders like inflammatory bowel disease, obesity, and cancer. MAGs also reveal functional genes of drug metabolism and resistance, setting the stage for personalized medicine. One example would be MAG-derived analyses of gut microbiomes, detecting pathways associated with host immune modulation and metabolic disorders.
Industrial Biotechnology
MAGs enhance the study of enzymes and pathways relevant to the industry. One example is the use of MAG identified enzymes involved in lignocellulose degradation for biofuels production. Genome mining of MAGs has also led to the discovery of novel antimicrobial compounds and secondary metabolites. These applications highlight the promising role of MAGs in solving major global issues like sustainable energy and antibiotic resistance.
Linking Evolutionary and Ecological Insights
MAGs provide insights into the evolutionary history and ecological roles of microorganisms. Using comparative genomics of MAGs, it will be able to trace the evolutionary origins of traits, like antibiotic resistance or symbiotic relationships. MAGs also retain the ability to provide insights into microbial interactions and niche specialization, essential for a holistic understanding of the communities in which they are found. Analyses of MAGs derived from coral reef microbiomes, for instance, have suggested roles of microbes in coral health and stress responses.
Directions and Innovations in the Future
Emerging technologies and methodologies in the MAG research field have already suggested ways to overcome current limitations and broaden the scope of applications.
Developments in Sequencing Techniques
Improved base-calling algorithms and the use of ultra-long-read sequencing will increase both the resolution and the completeness of MAGs. Moreover, these advancements will now allow for the reconstruction of more difficult and larger genomes, such as those of eukaryotic microbes. Continued cost reductions in sequencing, without a decline in the quality of the data, will increase access to MAG reconstruction.
Water Model Based Integration of Multi-omics Data
Integrating metagenomics with transcriptomics, proteomics, and metabolomics offers a more holistic perspective on microbial communities. Integrating multi-omics enables association of metagenome-assembled genomes (MAGs) with functional and phenotypic traits to provide insights into the function of microbes in ecological systems. This framework can be especially useful for interpreting microbial responses to environmental changes and their functional roles in generating ecosystem stability.
AI and Machine Learning
Use of machine learning algorithms to enhance metagenomic assembly, binning and annotation These tools improve genome reconstruction accuracy and minimize computational bottlenecks, thus streamlining the identification of new genomes and genes. AI-based binning approaches have shown the potential to reveal complex patterns present in these datasets, leading to enhanced high-quality MAG recovery from more complicated environments.
International Observatories and Databases
Vast amounts of metagenomic data are being generated by projects such as the Earth Microbiome Project and the Human Microbiome Project. Standardized workflows and open-access databases are needed to facilitate the utility of MAGs. These initiatives facilitate global collaboration and data sharing, accelerating discoveries of microbial genomics and ecology.
Case Study: Human Gut Microbiome MAGs Reconstruction
Background
The human gut microbiome is a complex microbial ecosystem with profound impacts on human health and disease. Despite its significance, a substantial proportion of its microbial diversity remains uncultured and poorly characterized. Researchers aimed to reconstruct high-quality MAGs from human gut metagenomes to better understand the composition, metabolic potential, and functional roles of these microbes.
Methods
- Sample Collection and Sequencing: Fecal samples from diverse human populations were collected to capture a broad representation of gut microbial diversity. Metagenomic sequencing was performed using Illumina technology for high-depth short reads and complemented with Oxford Nanopore for long reads to resolve complex regions.
- Assembly and Binning: Metagenomic reads were assembled using MEGAHIT for short-read assembly and Flye for long-read integration. Genome binning was performed using a combination of MetaBAT2 and MaxBin, leveraging sequence composition and differential coverage across samples to separate individual genomes.
- Quality Assessment and Refinement: CheckM was used to evaluate the completeness and contamination of MAGs. High-quality bins (>90% completeness, <5% contamination) were retained, and manual refinement was performed using Anvi'o. Functional annotations were carried out with Prokka and KEGG database mapping.
Results
- Novel Genome Recovery: Over 100,000 MAGs were reconstructed, including thousands of novel bacterial and archaeal genomes. A significant portion of these represented previously uncultured microbial taxa.
- Functional Insights: Key metabolic pathways were identified, including those involved in fiber degradation, short-chain fatty acid production, and amino acid biosynthesis. The study also uncovered microbial genes linked to drug metabolism and resistance, highlighting the microbiome's role in modulating therapeutic efficacy.
- Population-Level Analysis: Comparative analyses revealed microbial compositional differences across populations, shedding light on the impact of diet, geography, and lifestyle on gut microbiota diversity.
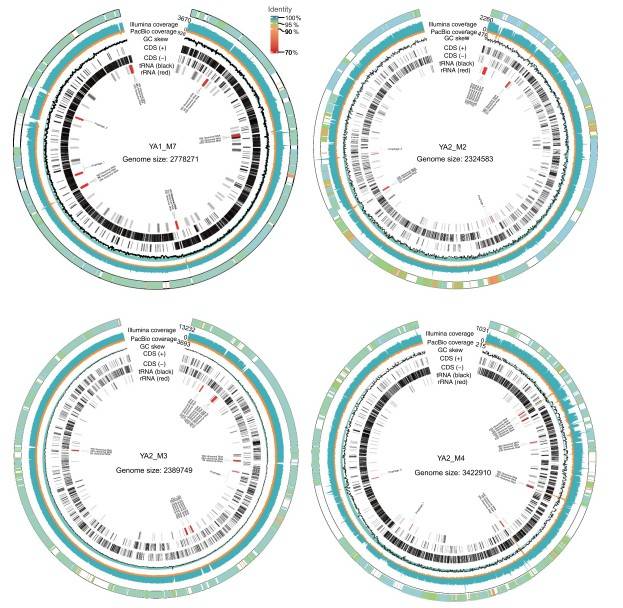 Genomic maps of four assembled complete (circularized, no gaps) MAGs (CMAGs) (Jin, H. et al 2021).
Genomic maps of four assembled complete (circularized, no gaps) MAGs (CMAGs) (Jin, H. et al 2021).
Conclusion
MAGs have opened the door to microbial genomics of the uncultivated majority of life. MAGs enable unprecedented insight into microbial diversity, ecology, and function by recovering genomes directly from environmental samples. While assembly and binning are not without their challenges, Genome Assembly approaches and toolsets are continuously evolving to provide higher-quality and more accessible MAGs. In this evolving landscape, MAGs will become ever more pivotal in solving global problems, such as those relating to environmental sustainability and human health, establishing their position in the genomic era.
References:
-
Goussarov, G., Mysara, M., Vandamme, P., & Van Houdt, R. (2022). Introduction to the principles and methods underlying the recovery of metagenome-assembled genomes from metagenomic data. MicrobiologyOpen, 11(3), e1298. https://doi.org/10.1002/mbo3.1298
- Jin, H., You, L., Zhao, F., Li, S., Ma, T., Kwok, L. Y., Xu, H., & Sun, Z. (2022). Hybrid, ultra-deep metagenomic sequencing enables genomic and functional characterization of low-abundance species in the human gut microbiome. Gut microbes, 14(1), 2021790. https://doi.org/10.1080/19490976.2021.2021790

 Sample Submission Guidelines
Sample Submission Guidelines


