In bioinformatics, genome assembly represents the process of putting a large number of short DNA sequences back together to recreate the original chromosomes from which the DNA originated. Sequence assembly is one of the basic steps after performing next generation sequencing, PacBio SMRT sequencing, or Nanopore sequencing. The established genome assembly can be submitted to databases such as European Nucleotide Archive, NCBI Assembly, and Ensembl Genomes. You can also browse these databases for genomic sequences done by other researchers.
Two types of genome assembly
There are two different types of genome assembly: de novo assembly and mapping to a reference genome (also known as reference-based alignment). De novo assembly refers to the genome assembly of a novel genome from scratch without the aid of reference genomic data. A reference genome or a reference assembly is a digital nucleic acid sequence database, acting as a representative example of a species’ set of genes. Once the reference genome is available, with its aid, the genome assembly becomes much easier, quicker, and even more accurate. Therefore, unless necessary, researchers choose the method of reference based alignment. Reference-based alignment has become the current standard in diagnostics.
Table 1. Reference-based alignment vs. de novo assembly.
|
Reference based alignment |
De novo assembly |
| Advantages |
- Good for SNV and small indels)
- Works for deletions and duplications by using coverage information
- A quick method to assembly the genome
- Hiding raw data limitations
- More tools to work with the results
- Easier annotation and comparison
|
- Dose not rely on a reference genome
- Used to search unknown genes/transcripts (such as transcripts with new intros, changed splice sites)
- Good for structural variations
|
| disadvantages |
- Requires a reference genome
- Limited by read length for feature detection
|
- Requires very high-quality raw data
- A slow method and requires high infrastructure
|
Evaluating an assembly
After the genome assembly, it is important to evaluate the quality of an assembly. The following table lists some of the important and commonly used assembly metrics. N50 is the most commonly used metric, which represents the smallest scaffold or contig length above which 50% of an assembly. It describes the “completeness” of an assembly.
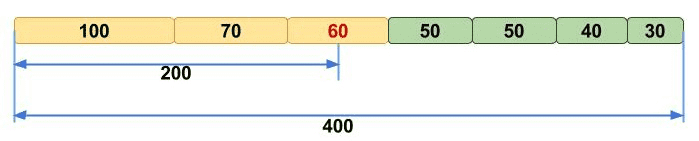 Figure 1. The calculation example of N50.
Figure 1. The calculation example of N50.
Table 2. Some common statistics used in evaluating the quality of an assembly
| Metrics |
Description |
| N50 |
N50 means, half of the genome sequence is larger than or equal the N50 contig size (↑). |
| NG50 |
The length of the scaffold at which 50% of the genome length is covered (↑). |
| Coverage |
If 90% of the bases have at least 5X read coverage, the genome is considered accurate (↑). |
| N90 |
An assembly is considered to have continuity provided its N 90 > 5 Kb (↑). |
| Average contig length |
The average contig length should be longer than 5000 bases (5 Kb) (↑). |
| Number of genes |
If an assembly that identifies most of the known genes is considered the better assembly (↑). |
| Number of gaps |
The gaps in an assembly decreases the quality (↓). |
| Validity |
An assembly can be validated by the reference sequence (↑). |
Notice that an ↑ indicates that higher is better and a ↓ implies that less is better.
Factors affecting genome assembly results
In addition to the process of genome assembly, the following issues can strongly affect the quality of genome assembly. Paired-end sequencing and long-read technology are two strategies to improve the quality of genome assembly.
1. Properties of the genome
The properties of the genome may affect the genome assembly.
- Genome size. The bigger the genome is, the more data is needed. Therefore, before ordering sequence data, you need to estimate the genome size, which may be inferred by investigating the genome size of closely related species.
- Repeats. Amount and distribution of repeated sequences in a genome largely influence the genome assembly results. This can lead to misassemblies and an incorrect estimate of the size of the repeats.
- Heterozygosity. If the genome is highly heterozygous, sequence reads from homologous alleles can be too different to be put together, probably leading to more fragmented assemblies or creating doubt about the homology of the contigs.
- Ploidy level. If possible, it is better to sequence haploid tissue, avoiding problems caused by heterozygosity.
- GC-content. Inhomogenous GC content can cause a problem for Illumina sequencing, resulting in low coverage in these regions.
2. Nucleic Acid Extraction
For the DNA isolation or RNA isolation, here are a couple of things need to be aware of: DNA/RNA integrity, DNA/RNA purification, sufficient DNA/RNA amount, etc. Compared with resequencing, de novo sequencing requires superior nucleic acid. The most important nucleic acid quality parameters for NGS are chemical purity and structural integrity.
3. Sequencing methods
The determination of sequencing methods is an important factor that influences the cost and success of a genome assembly. NGS has been applied to in many remarkable projects such as the 1,000 Genomes Project and the Human Microbiome Project. However, some important genes of interest cannot be assembled correctly, mainly due to the interruption of repeat elements. Third generation sequencing is a promising solution to this problem based on long reads that span the repetitive regions.
4. Raw data processing
Although there are assembly tools that prefer dealing with the raw data, including potential adapter sequences, we highly recommend that researchers study the manual to determine whether the program requires quality-trimmed data or not. If data trimming is required, it would be necessary to omit poor quality data by trimming low quality read ends and filtering of low quality reads. Multiple tools are available for this purpose, such as PRINSEQ32 and Trimmomatic33.
References:
- Wajid B, Serpedin E. Do it yourself guide to genome assembly. Briefings in functional genomics, 2014, 15(1): 1-9.
- Victoria D D A, Erik H, Lieven S, et al. Ten steps to get started in Genome Assembly and Annotation. F1000Research, 2018, 7.
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


