Conventional genome assembly is a key computational task in genomics, where the assembler reconstructs the complete genome from a collection of short summaries of DNA or RNA sequence.This process underpins much of biological research, from questions of gene function, evolutionary research, and forensic applications. Genome assembly is a first step in many genomic studies, and the quality of the assembly informs its use for variant calling and phylogenetic inference. Genome assembly is facilitated and optimized by specialized tools and algorithms designed to address these challenges, including aspects like repetitive sequences, sequencing errors, and the sheer amount of raw data produced by high-throughput sequencing technologies.
Genome assembly has evolved along with genome sequencing technologies (Sanger, Illumina, PacBio, and Oxford Nanopore) and associated assembly methodologies. These resulting tools have allowed for the assembly of genomes ranging from small viral genomes to large-eukaryote genomes that are typically highly repetitive and polyploid. Every tool has its own given limitations and advantages, which also holds true for given types of data, as well as levels of complexity of genomes and research aims. This article will provide an overview of the major classes of genome assembly tools, what they do, and how they have evolved in the genomics landscape.
Genomic tools for genome assembly
Genome assembly tools can generally be divided by type of the sequencing data on which they apply and by the assembly strategy they follow. In consonance, the above categories cover a biologically relevant spectrum of challenges — accuracy, scalability and assembly of more complex genomes. To address the issues emerged in the process of de novo genome assembly, more and more tools are developed.
Service you may interested in
Resource
Short-read Assembly Tools
A short-read sequencing platform is Illumina, which creates single reads of extremely high accuracy, typically 50 to 300 base pairs long. These short reads are ideal for covering areas deeply as well as finding small variants, but they do not resolve long-range genomic structures or repetitive areas.
- SPAdes: SPAdes is the most widely used tool for small genome assembly. Its De Bruijn graph-based approach enables it to handle short-read datasets swiftly. SPAdes: for microbial genomes, metagenomes, and transcriptome assemblies. Due to its strong error-correcting codes and its use of an iterative assembly process, the device achieves very low error rates, making it a popular choice for implementations where fidelity is desired.
- Velvet: Velvet was one of the first tools built for the short-read assembly and is still a very readable choice for moderately complex genome projects. Building De Bruijn graphs with it is memory-economical—computational length is sacrificed for the sake of assembly accuracy. Velvet especially shines with datasets that have constant coverage.
- SOAPdenovo: Designed for genomes of large projects, SOAPdenovo excels in assembling plant and animal genomes from short-read data. It uses parallel computing techniques to handle the computational demands of large datasets and permits researchers to assemble genomes with long repeat regions as long as sequencing depth is adequate.
Long-read Assembly Tools
Long-read sequencing platforms (PacBio and Oxford Nanopore) produce reads in tens of kilobases. These reads are particularly well-suited to addressing repetitive sequences, structural modifications and other challenging areas of genomes that short-read data fails to tackle.
- Canu: The Canu assembler is an excellent assembler for high-error long-read data. Canu uses an Overlap-Layout-Consensus (OLC) algorithm that aligns reads, finds overlaps, and arranges reads creating very contiguous assemblers. It is particularly good for constructing the genomes of large, complex organisms — even repeat-rich organisms — and has opened the door to reference-quality assembly of the genomes of plants and animals.
- Flye: This assembler is optimized for speed and for assembling genomes given noisy long-read data. Its error tolerant algorithms allow it to reconstruct microbial and eukaryotic genomes at both high efficiency. Flye's fast workflows enable a spectrum of applications from academic research to industrial applications requiring accelerated turnaround.
- Shasta: This technology is directed toward ultra-long reads and enables the quick processing of large genomes with good computational efficiency featuring state of the art algorithms for read error correction and assembly layout creation to produce very contiguous assemblies for large-scale projects such as human genomes.
Hybrid Assembly Tools
Hybrid method assembly algorithms exploit the benefits from both short reads and long reads sequencing reads. This moves them to manufacture the strong solution to assemble difficult genomes organizing as both the precision of short read and the degree of long read.
- MaSuRCA:Software to attempt an assembly with an Illumina short reads and a PacBio or Nanopore long reads. Its algorithms iteratively align and merge reads to promote contiguity and accuracy while resolving repetitive regions. MaSuRCA is particularly good for large repetitive genomes; plant and amphibian genomes fit this descriptor.
- Unicycler: Designed around bacterial genome assembly, Unicycler supports both short- and long-read data for complete, circularized assemblies It has become the tool of choice for microbial genomics because of its fidelity with respect to plasmids and small genomes.
- SPAdes (Hybrid Mode): Extensions to SPAdes include hybrid datasets that integrate multiple sequencing platforms for improved assembly quality. This is particularly helpful for metagenomic projects that are combining several sequencing technologies.
Genome Assembly Tools Functions
There is a variety of genome assembly tools to accommodate the challenges of sequencing data inherent in the process. Such characteristics are important for preserving genome assembly completeness and accuracy.
- Error Correction: Sequencing errors decrease assembly quality, especially in datasets created by high-error platforms, such as Nanopore. Tools like Pilon (short reads) and Racon (long reads) further polish assemblies by detecting and resolving mismatches, greatly improving base accuracy.
- Scaffolding: Scaffolding connects contigs into larger, ordered structures using other data, like mate-pair reads or long reads. SPACEs and LINKs leverages this spatial information to generate scaffolds that reflect the chromosomally ordered arrangement of sequences, thus increasing the contiguity and accuracy assemblies.
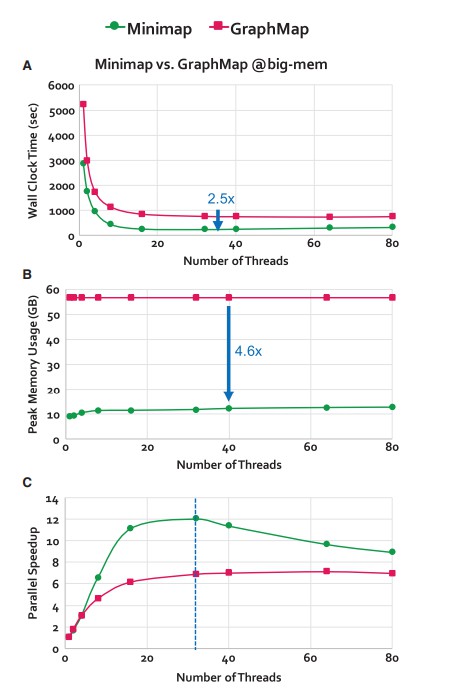 Scalability results of Minimap and GraphMap (Senol Cali et al. 2019).
Scalability results of Minimap and GraphMap (Senol Cali et al. 2019).
- Polishing: Polishing tools correct last assembly residual errors to fulfill the highest accuracy in assemblies. For example, Nanopolish and Arrow are specifically designed to polish long-read-based assemblies, and Pilon is one of the most popular tools for polishing Illumina-based datasets.
- Repeat Resolution: Repeats can be one of the most difficult features to resolve in genome assembly, and are especially present as a challenge within eukaryotic genomes. Many assemblers, including Canu and Flye, contain algorithms for identifying and resolving repetitive regions, allowing them to reconstruct full sequence, as well as the contiguous sequence.
Genome Assembly Tools: Trends and Advances in Software
Genome assembly tools are also evolving with sequencing technologies to meet new challenges and leverage new possibilities. Key advancements include:
- Ultra-long Read Assemblies: As ultra-long reads (greater than 1 Mb in length) become more accessible, tools have emerged that assemble genomes with an unprecedented level of continuity. These tools allow researchers to deconvolute previously intractable regions, such as centromeres and telomeres, shedding light on aspects of chromosomal architecture and function.
- Artificial Intelligence and Machine Learning: Machine learning algorithms are being used to enhance genome assembly workflows, aimed at maximizing error correction, repeat resolution and structural variance detection. By integrating AI-driven tools, assembly processes are enhanced in speed and accuracy, especially in more complex regions and larger genomes. Details can refer to our article "Genome Indexing in Bioinformatics: Unpacking the Genome".
- Cloud-based Pipelines: The era of cloud computing offers scalable and a less expensive means to process data in the genome assembly. These platforms allow researchers to build large, complex genomes without significant local computational infrastructure.
- End to End Automation: Pipelines with an integrated system that can automate genome assembly, annotation, and visualization are gaining prevalence. Such systems facilitate workflow, minimize human interference, and improve the reproducibility of genomic analyses.
Genome Assembly Tools & It's Significance In Present Day
Genome assembly tools have become required instruments in genomics, enabling discoveries in numerous fields. They form a basis for applications in medical research, agriculture, evolutionary biology, and more.
Background
One of the biggest scientific feats of the 20th century was the human genome project (HGP), which sought to sequence and assemble the complete human genome. It was initiated in 1990 and relied on early technologies and computational methods to work. The human genome assembly was a pivotal moment in the field of genomics, offering a reference map for elucidating human biology and disease.
Methods
- Sequencing: The early phases of the project employed Sanger sequencing, which produces high-quality reads but is time-consuming and expensive. BAC (bacterial artificial chromosome) clones that overlapped were sequentially run to sectionalize the genome into smaller chunks.
- Assembly Strategies: Hierarchical shotgun sequencing approach was used. BACs were used to clone large DNA fragments, which were then digested into smaller fragments and sequenced. Computational algorithms aligned and assembled these fragments into contiguous sequences (contigs) and scaffolds.
- Validation: The quality of the assembly was validated by comparing with known genetic markers and by mapping against physical maps of the genome. Subsequent stages incorporated high-throughput sequencing data to fill in missing information and resolve ambiguities.
Results
The HGP produced a draft of the human genome in 2001 that accounted for more than 90% of the genome and that was accurate down to the level of bases. In 2003, there released a nearly complete version with around 99% coverage and minimal gaps. The assembled genome was 3 billion base pairs with 20,000–25,000 putative protein-coding genes. The Human Genome Project is widely regarded as a landmark achievement, which enabled many advances, such as identification of disease causing genes, development of targeted therapies, and exploration of human evolutionary history.
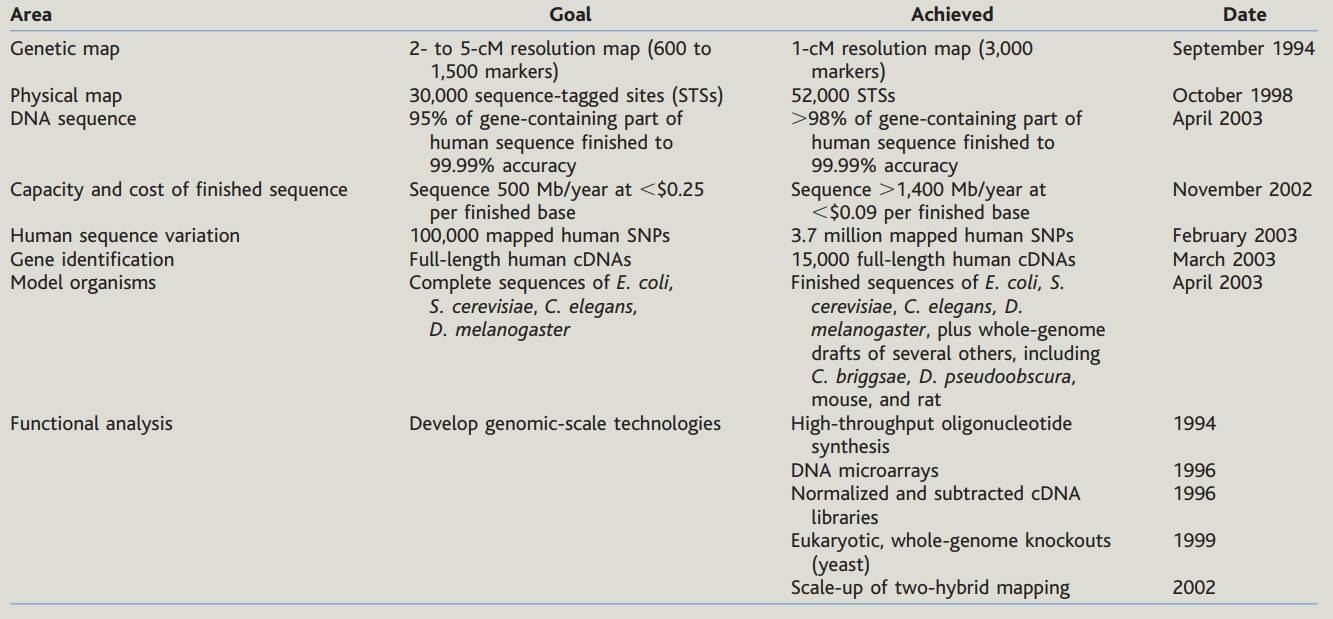 HGP goals and dates of achievement (Collins, F. S. et al 2003)
HGP goals and dates of achievement (Collins, F. S. et al 2003)
Conclusion
Tools for genome assembly have revolutionized the field of genomics, enabling researchers to reconstruct high-quality genomes with ever-increasing accuracy and efficiency. Specific datasets and research objectives can also encourage or hinder efficient genome assembly. With sequencing technologies and computational methods continuing to evolve, these tools are set to play an even greater role in developing our concept of life's genetic blueprint.
References:
- Senol Cali, D., Kim, J. S., Ghose, S., Alkan, C., & Mutlu, O. (2019). Nanopore sequencing technology and tools for genome assembly: computational analysis of the current state, bottlenecks and future directions. Briefings in bioinformatics, 20(4), 1542–1559. https://doi.org/10.1093/bib/bby017
- Collins, F. S., Morgan, M., & Patrinos, A. (2003). The Human Genome Project: lessons from large-scale biology. Science (New York, N.Y.), 300(5617), 286–290. https://doi.org/10.1126/science.1084564

 Sample Submission Guidelines
Sample Submission Guidelines


