
 Sample Submission Guidelines
Sample Submission Guidelines
With CD Genomics' extensive proficiency in next-generation sequencing (NGS), we are now pleased to offer Double Digest Restriction-site Associated DNA (ddRADseq) services for comprehensive genome-wide SNP discovery, even in the absence of prior genomic sequence information. ddRADseq facilitates the sequencing of genome-scale data from non-model species, enabling cost-effective development of extensive datasets that encompass broad taxonomic and geographic sampling.
What is ddRAD Sequencing
ddRADseq has gained considerable popularity among molecular ecologists for the development of novel SNP markers utilizing NGS platforms. This method leverages the cut-site specificity of restriction endonuclease enzymes to generate library fragments from distinct genomic regions that are conserved across individuals of the same species. This conservation enables the sequencing and comparative analysis of identical genomic regions across different individuals. Consequently, ddRADseq facilitates the rapid and efficient development of a substantial number of genetic markers.
What is the Principle of ddRAD Sequencing
ddRADseq is an enhanced variation of the traditional RAD sequencing protocol, specifically designed for SNP discovery and genotyping. Instead of fragment shearing, ddRAD-seq incorporates a secondary restriction digestion, thereby enhancing both the tunability and accuracy of the size-selection step. In brief, the process begins with the digestion of genomic DNA using a restriction enzyme, followed by the ligation of a barcoded P1 adaptor to the resulting fragments. These adaptor-ligated fragments from various samples are subsequently pooled, and a second restriction enzyme is applied for further digestion. The digested fragments are then subjected to size-selection and purification. Thereafter, P2 primers are ligated, and the fragments are amplified. Ultimately, sequence data are analyzed to assess and score genetic variations within the samples of interest.
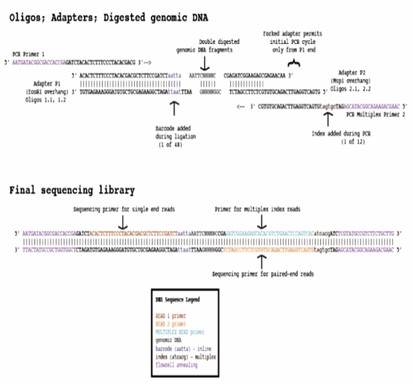
What are the Advantages of ddRAD Sequencing Service
- Reusability of Samples: Multiple samples can be reused, maximizing resource efficiency.
- Efficient Workflow: The library preparation process is simpler and faster, streamlining the overall experimental procedure.
- Consistent Fragment Length: Ensures uniform fragment lengths across samples at identical digestion sites, improving accuracy.
- SNP Density Flexibility: Offers flexibility in SNP density, allowing customization based on specific research needs.
- High Sequence Coverage: Achieves high sequence coverage, enhancing data reliability.
- Cost-Effectiveness: Lowers experimental costs while maintaining robust data output.
- No Reference Genome Required: Enables SNP discovery without the need for a reference genome.
- Advanced Multi-Technology Platform: Provides dependable data results, meeting both reliability and cost-efficiency requirements.
- Technical Support: Comprehensive support services cater to all project needs, ensuring customer satisfaction.
- Streamlined Experimental Process: Simplifies the experimental process and ensures rapid turnaround. Multi-locus projects can be conducted in multiplexed assays with typing completed in a single tube.
- Real-Time Project Updates: Offers real-time project progress updates, allowing customers to track each step clearly.
- Customized Protocols: Flexibility to tailor experimental protocols to project-specific needs, with expert customization for optimal results.
Applications of ddRAD Sequencing
- Population Genetics
- Genome-Wide Association Studies (GWAS)
- Conservation Biology
- Evolutionary Biology
- Genomic Mapping
- Breeding Programs
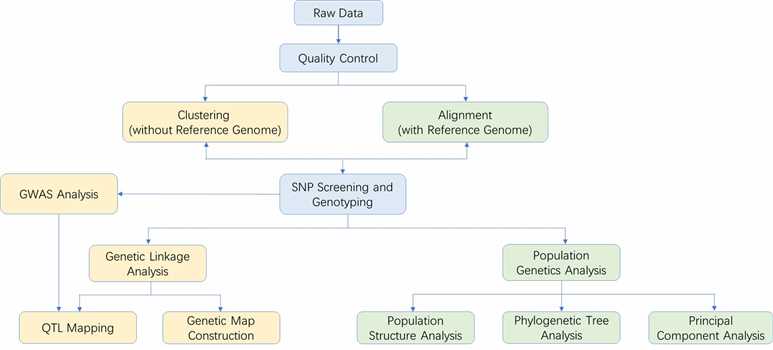
ddRAD Sequencing Workflow
The workflow of ddRAD Sequencing at CD Genomics involves several key steps:

Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis
We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in ddRAD-seq for your writing (customization)
CD Genomics delivers a complete ddRAD-seq service, handling everything from initial DNA quality checks to detailed SNP analysis. We also offer customized solutions designed to fit your specific project needs. For further information or to discuss your requirements, please contact our team.
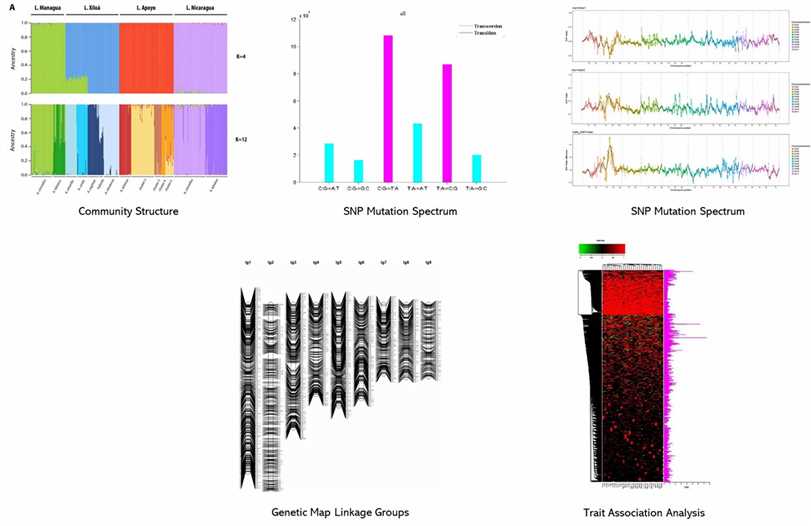
Partial results are shown below:
1. How long does it take to get results from ddRAD-seq?
The typical turnaround time for ddRAD-seq results varies depending on project size and complexity but usually ranges from 4 to 8 weeks.
2. Can we customize the ddRAD-seq service for specific needs?
Yes, we offer tailored solutions to meet unique project requirements, including custom protocol adjustments and data analysis options.
3. What types of samples are suitable for ddRAD-seq?
ddRAD-seq can be applied to various sample types, including blood, tissue, and other genomic DNA sources from plants, animals, and microorganisms.
4. Can ddRAD-seq be used without a reference genome?
Yes, ddRAD-seq does not require a reference genome, making it suitable for species where reference genomes are not available.
5. How to choose the appropriate reduced-representation genome technology?
When selecting the appropriate simplified genomics technology, consider the following four factors to design your research strategy:
Reference Genome
- With Reference Genome: Using a reference genome helps reduce errors due to homologous or repetitive sequences, facilitates LD analysis, selection analysis, and GWAS. This is suitable for conventional RAD sequencing and PE151 sequencing.
- Without Reference Genome: ddRAD-seq is recommended.
Sequencing Strategy
- Double Digest: For short fragments, paired-end (PE) sequencing may result in significant adapter overlap and is not ideal for long read lengths; single-end (SE) sequencing is recommended for short fragments.
- Long Fragments: Long reads can detect more variant information but require sufficient coverage.
- Short Reads: Improves sequencing depth per restriction site, enhancing SNP detection accuracy.
- Non-Reference Species: SE sequencing is recommended to avoid data wastage.
Number of Markers
- High Marker Density: Conventional RAD sequencing is suitable for analyses requiring high-density markers.
- Complex Genomes and Large Sample Sizes: GBS sequencing is ideal for complex genomes and large sample volumes.
PCR Amplification Artifacts
- PCR Bias: May lead to heterozygous sites being misidentified as homozygous or introduce errors. Conventional RAD sequencing can remove PCR duplicates using fragment sequence information, while GBS and ddRAD-seq cannot.
ddRAD sequencing-based genotyping for population structure analysis in cultivated tomato provides new insights into the genomic diversity of Mediterranean 'da serbo' type long shelf-life germplasm Horticulture Research
Journal: Horticulture Research
Impact factor: 5.404
Published: 01 September 2020
Background
Tomato (Solanum lycopersicum L.) stands as a vital economic vegetable crop with global cultivation. One can trace the beginnings of tomato domestication to the Andean region of South America, from where it spread throughout the Americas. During the 16th century, tomatoes were introduced to Europe, with Spain and Italy as major entry points. This introduction sparked further domestication efforts, contributing to the emergence of substantial local genetic diversity. Consequently, the Mediterranean Basin in Europe is recognized as a secondary center for the diversification of tomatoes. In the context of this investigation, the authors harnessed ddRAD-seq technology to methodically explore the genetic diversity present within various tomato varieties.
Materials & Methods
Sample Preparation
- Tomato
- DNA extraction
Method
- RAD-seq genotyping
- HiSeq2500 instrument
- SNP call
- Filtering and marker classification
- Population structure
- Genetic diversity
- Phenotypic analysis
Results
In this investigation, the genetic diversity of 288 tomato specimens was examined, encompassing 152 samples of the long shelf life (LSL) variety 'da serbo,' primarily originating from Italy and Spain, as well as other common varieties from diverse countries. Beyond the LSL trait, 'da serbo' varieties also display stress tolerance characteristics. In order to do non-parametric hierarchical clustering and model ancestral population structure, the study found 32,799 high-quality single nucleotide polymorphisms (SNPs). six distinct genetic subpopulations were delineated, effectively segregating the majority of 'da serbo' varieties, reflecting population substructure related to both varietal type and geographical origin. Linkage disequilibrium (LD) exhibited rapid decay within a genomic span of less than 5kb. Investigation of SNPs featuring low-frequency alleles (MAF) in 'da serbo' materials unveiled a high-frequency mutation in genes associated with stress tolerance, such as CTR1 and JAR1, which are implicated in fruit ripening. Finally, leveraging a selection of 58 core materials representing a significant portion of the genetic diversity, essential traits were further developed. The genetic signatures of the 'da serbo' germplasm, which was cultivated with selection in the Mediterranean Basin, are revealed in this work. Furthermore, it provides new perspectives on the long-lived "da serbo" germplasm, establishing it as a rich source of stress-tolerance genes.
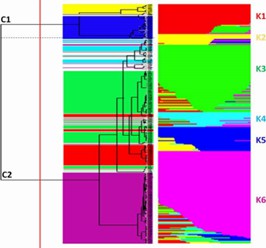 Fig. 1 Estimate of genetic diversity in 288 tomato accessions using ddRAD.
Fig. 1 Estimate of genetic diversity in 288 tomato accessions using ddRAD.
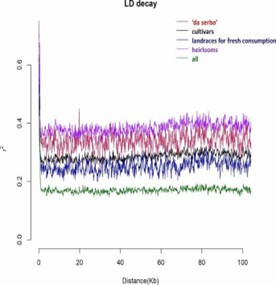 Fig. 2 Linkage disequilibrium (LD) decay and comparison.
Fig. 2 Linkage disequilibrium (LD) decay and comparison.
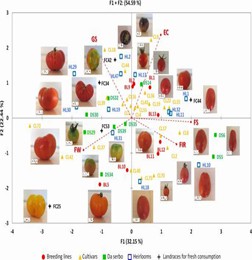 Fig. 3 Loading plot of the first (F1) and second (F2) principal components showing the variation for main fruit traits in accessions of the mini-core set developed from ddRAD SNP data of 288 cultivated tomato genotypes.
Fig. 3 Loading plot of the first (F1) and second (F2) principal components showing the variation for main fruit traits in accessions of the mini-core set developed from ddRAD SNP data of 288 cultivated tomato genotypes.
Conclusion
Using ddRAD-seq and the latest tomato genome, the authors identified 2,297 new target genes with SNPs in the Mediterranean 'da serbo' gene pool. This study reveals a geographic distinction among Mediterranean landraces and highlights SNPs in novel gene regions related to stress and hormone pathways. A mini-core collection of diverse genotypes was provided for breeding, offering a valuable gene reservoir for future precision breeding and genome-wide association studies.
Reference
- Esposito, S., Cardi, T., Campanelli, G. et al. ddRAD sequencing-based genotyping for population structure analysis in cultivated tomato provides new insights into the genomic diversity of Mediterranean 'da serbo' type long shelf-life germplasm. Hortic Res 7, 134 (2020).
Here are some publications that have been successfully published using our services or other related services:
Use of biostimulants for water stress mitigation in two durum wheat (Triticum durum Desf.) genotypes with different drought tolerance
Journal: Plant Stress
Year: 2024
The Restriction-Modification Systems of Clostridium carboxidivorans P7
Journal: Microorganisms
Year: 2023
In the land of the blind: Exceptional subterranean speciation of cryptic troglobitic spiders of the genus Tegenaria (Araneae: Agelenidae) in Israel
Journal: Molecular Phylogenetics and Evolution
Year: 2023
Genetic Modifiers of Oral Nicotine Consumption in Chrna5 Null Mutant Mice
Journal: Front. Psychiatry
Year: 2021
A high-density genetic linkage map and QTL identification for growth traits in dusky kob (Argyrosomus japonicus)
Journal: Aquaculture
Year: 2024
Genomic and chemical evidence for local adaptation in resistance to different herbivores in Datura stramonium
Journal: Evolution
Year: 2020
See more articles published by our clients.


