Antibiotic Resistance Genes (ARGs) and Their Impact on Public Health
Antibiotic resistance genes (ARGs) are genetic elements located within bacterial or other microbial genomes that confer the ability to withstand the effects of antibiotics. These genes encode a variety of proteins or other molecular mechanisms that enable bacteria to develop resistance to antibiotic treatments.
ARGs are increasingly being recognized as emerging biological pollutants. The resistance they induce poses one of the most significant challenges to contemporary medicine and represents a critical public health concern. Antibiotic resistance hampers the effective treatment of bacterial infections in clinical settings and contributes to the spread of epidemics and increased mortality rates.
According to data obtained from the European Union and European Economic Area countries, antibiotic-resistant bacteria (ARB) were responsible for 671,689 infections in 2015, leading to over 33,000 deaths annually.
The widespread dissemination of ARGs in the environment exacerbates their threat as contaminants. Extensive use of antibiotics in human medicine, veterinary practices, and agriculture results in their continuous release into the environment. Concurrently, ARGs infiltrate various environmental settings, including clinical (hospitals, clinics) and veterinary environments, the human gastrointestinal microbiome, as well as wastewater treatment facilities and natural habitats, such as soil, water, and air.
Mobile genetic elements (MGEs), such as plasmids, transposons, and integrons, have been demonstrated to be strongly associated with the mobility and spread of ARGs. Establishing the link between ARGs and MGEs is crucial for understanding and mitigating the dissemination of antibiotic resistance.
Methods and Tools in Bioinformatics for ARGs Research
The primary objective of bioinformatics in the study of ARGs is the development of analytical workflows designed to precisely detect resistomes, which are extensions of resistance gene collections. Subsequently, these workflows aid in the accurate prediction of antimicrobial susceptibility profiles, encompassing the phenotypic range and susceptibility of antimicrobial resistance (AMR), as well as information pertaining to ARG hosts, based on genomic and metagenomic data.
A myriad of bioinformatics software tools and databases related to ARGs has been developed to date. However, the annotation and management of these tools and databases often lack consistency, with each tool and database emphasizing different focal areas and exhibiting varying scopes of prediction.
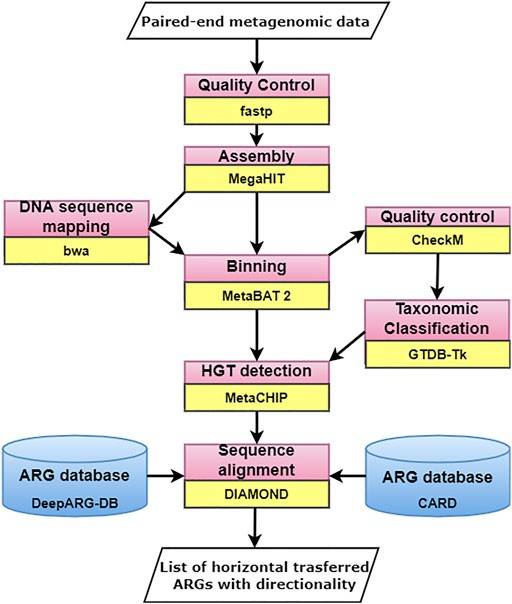 Workflow for Analyzing ARGs in Metagenomic Sequencing Data (Badhan Das et al,.2022)
Workflow for Analyzing ARGs in Metagenomic Sequencing Data (Badhan Das et al,.2022)
Currently, two principal computational workflows are utilized for the identification and characterization of ARGs present within microbial communities using metagenomic sequencing data: assembly-based analysis of contigs and alignment-based analysis of raw reads .
However, most methodologies applied to sequences derived from metagenomic data-whether raw reads or contigs-are generally unable to precisely attribute these sequences to specific species origins. Consequently, the analysis scope is often limited to ARG families that can be identified based solely on homology. This approach often lacks the capacity to analyze resistance determinants that are highly environment-dependent, such as those arising from point mutations.
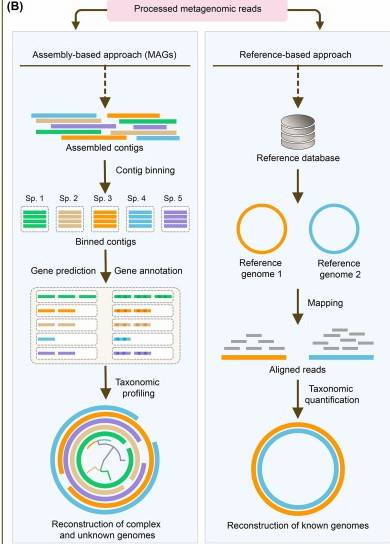 A schematic contrast between assembly-based and reference-based approaches on metagenomic sequencing data. (Chao Yang et al,.2021)
A schematic contrast between assembly-based and reference-based approaches on metagenomic sequencing data. (Chao Yang et al,.2021)
Services you may interested in
Identification of Resistance Genes: Assembly-Based vs. Read-Based Approaches
Assembly-Based Contig Analysis
In the assembly-based approach, sequence reads are partitioned into shorter overlapping subsequences (k-mers) using De Bruijn graph-based assembly programs such as SPAdes, Velvet, ABySS, or SOAPdenovo. These k-mers are organized into a network, where the optimal path is identified to reconstruct the gene sequences. Once assembly is complete, protein-coding regions on the contigs are predicted, and resistance genes within the assembled genomic or metagenomic contigs are identified and annotated. This process involves comparing the contigs against reference databases using similarity search tools like BLAST, USEARCH, or DIAMOND.
However, in metagenomic data, uneven sequencing coverage across different organisms can complicate detection and assembly. Therefore, algorithms designed for single-genome assembly may not be directly applicable to metagenomic assembly. Commonly used metagenomic assembly tools include IDBA-UD, MEGAHIT, MetaSPAdes, MetaVelvet, and CAMI.
Read-Based Analysis
Antibiotic resistance genes in a sample can also be identified without the need for genome assembly. This approach involves aligning the sequence reads to a reference database or genome using pairwise alignment tools such as Bowtie2 or BWA. Alternatively, the sequence reads can be fragmented into k-mers, which are then mapped to the reference database.
Advantages and Disadvantages of Both Approaches
Both approaches have distinct advantages and limitations. While assembly-based methods may lose some information, they allow for the identification of protein-coding genes and the investigation of upstream and downstream regulatory elements. In contrast, read-based analysis lacks information regarding the location of upstream and downstream factors of identified resistance genes.
| Method | Characteristics |
|---|---|
| Assembly-Based Contig Analysis | (1) High computational cost and time, especially for large and complex communities; (2) Identification of resistance genes, both known and novel, with low similarity to reference databases, but requires high genomic coverage; (3) Ability to capture regulatory elements, mobile element sequences, and gene backgrounds. |
| Read-Based Analysis | (1) Fast with low computational demands, suitable for analyzing large datasets; (2) Identification of resistance genes depends on the completeness of the reference database for the organisms analyzed; (3) Loss of gene background and nearby genes; potential for false positives due to misalignment. |
Currently, there is no consensus on which method is superior, and the choice between these two approaches should be guided by sequencing type, available computational resources, and specific research objectives.
Tools for Detection and Common Databases for ARGs
Detection and Prediction Tools
| Tool Name | Description |
|---|---|
| ResFinder | A tool for detecting acquired resistance genes from fully or partially sequenced bacterial isolates. |
| ARG-ANNOT | A tool for comparing query sequences against the ARG-ANNOT database. |
| RGI | (1) Compares query sequences against the CARD database. (2) Uses curated resistance gene detection models to predict intrinsic, specific, and mutation-acquired resistance genes. |
| ARGs-OAP (v2) | An online tool for quickly annotating and classifying antibiotic resistance gene-like sequences from metagenomic data using BLASTX against the SARG database. |
| ARIBA | A tool for rapid resistance gene typing from sequencing fragments using managed public databases. |
| PointFinder | A web tool to identify resistance gene and chromosomal point mutations associated with bacterial pathogens. |
| NCBI-AMRFinder | A tool that uses NCBI's antimicrobial resistance gene database and collected sequence feature profiles to identify resistance genes. |
Tools for Read-Based Analysis
| Tool Name | Description |
|---|---|
| SRST2 | Uses Bowtie2 to align reads to a custom reference database for predicting antibiotic resistance genes in samples. |
| SEAR | A cloud-based alignment tool for rapid detection of resistance genes from sequencing fragments with a web interface. |
| ShortBRED | A tool that reflects protein families in metagenomic data using short peptide marker sequences. |
| PATRIC | A unique resource for researching resistance genes. |
| SSTAR | A tool to identify novel and existing trimmed resistance gene alleles from whole-genome data. |
| KmerResistance | Splits raw reads into k-mers and maps them to identify co-occurrences, predicting resistance genes and associated species. |
| GROOT | A software tool for analyzing and studying resistance gene clusters by mapping metagenomic reads to reference gene sets. |
| DeepARG | A deep learning method for predicting resistance genes from metagenomic data. |
Databases
With the rapid growth of ARG data in recent years, the need for improved data management, analysis, and access has increased, requiring more comprehensive databases. The naming of ARGs itself presents challenges, with frequent synonym use, conflicts in names, and inconsistency between nucleotide- and protein-based names. Furthermore, various ARG databases informally exchange information about AMR management, naming, and classification, leading to some disorganization. Additionally, no ARG database is complete, and the movement and mutation of ARGs between pathogens contribute to the vastness of data, with mutation data management being a particularly challenging task.
| Database Name | Description |
|---|---|
| CARD | A comprehensive database managed by the Antibiotic Resistance Ontology (ARO) that provides information on resistance genes and their mechanisms, along with a Resistance Gene Identifier (RGI) tool. |
| ResFinder | Detects antibiotic resistance genes in fully or partially sequenced bacterial isolates, using PointFinder to identify chromosomal mutations. |
| Resfams | A database of antibiotic resistance proteins based on hidden Markov models (HMM) used for identifying resistance gene functions. |
| ARDB | The first manually curated database that collects resistance gene information, containing at least 4500 resistance gene sequences. Data has been integrated into CARD since 2009. |
| MEGARes | Combines multiple databases (CARD, ARG-ANNOT, ResFinder) to avoid sequence redundancy for high-throughput screening and data analysis. |
| NDARO | A comprehensive collection derived from multiple databases (CARD, Lahey, ResFinder, Pasteur Institute β-lactamases) covering 4500 resistance gene sequences. |
| ARG-ANNOT | A database containing over 1800 resistance gene sequences collected from scientific literature and online resources, including chromosomal point mutation data related to resistance genes. |
| Mustard | Includes 6095 resistance gene determinants from 20 families and contains a resource for identifying functional metagenomics of resistance genes. |
| FARME Database | A database of functionally screened microbial sequences for resistance across different habitats in various functional metagenomic studies. |
| SARG | A hierarchical database derived from ARDB, CARD, and NCBI-NR, encompassing over 12000 resistance genes and 189 resistance gene subtypes using HMM profiles. |
| Lahey List of β-lactamases | A collection of known β-lactamases and newly designated enzymes with names assigned to novel variants. |
| BLDB | A manually curated database of antibiotic resistance enzymes categorized by class, order, and family. |
| Laced | A database that includes TEM and SHV β-lactamases and their variants. |
| CBMAR | A database for the identification and characterization of novel β-lactamases based on the Ambler classification. |
| NCBI BioProject | A database of genomic, functional genomics, and genetics research datasets and their results. |
Species-Specific Databases
| Database Name | Description |
|---|---|
| MUBII-TB-DB | A database of mutations associated with antibiotic resistance genes in Mycobacterium tuberculosis. |
| u-CARE | A user-friendly and comprehensive database of resistance genes in Escherichia coli. |
Many ARG databases have not been updated or actively managed for some time. Currently, CARD (Comprehensive Antibiotic Resistance Database) stands out as one of the most comprehensive and well-maintained databases, with many of its entries experimentally validated. CARD provides reference DNA and protein sequences, detection models, and bioinformatics tools based on the molecular mechanisms of AMR. It incorporates the Antibiotic Resistance Ontology (ARO), which is designed to categorize resistance-related data.
The ARO is structured into three primary branches:
1. Antibiotic Resistance Determinants (ARO:3000000)
2. Antibiotic Molecules (ARO:1000003)
3. Antibiotic Resistance Mechanisms (ARO:1000002)
4. The most recent version of CARD includes over 6000 ontology terms, many of which are supported by numerous peer-reviewed studies.
CARD has also developed the Resistance Gene Identifier (RGI) software (v5.0), which utilizes four types of models to predict resistance groups:
- Protein Homology Model: Detects AMR gene homologs using BLASTP or DIAMOND.
- Protein Mutation Model: Accurately distinguishes between susceptible intrinsic genes and acquired mutations that confer AMR, based on a curated single-nucleotide polymorphism (SNP) matrix.
- rRNA Mutation Model: Detects mutations in rRNA target sequences associated with resistance.
- Protein Overexpression Model: Identifies mutations that cause overexpression of efflux pump subunits, which are often associated with AMR.
Consequently, RGI + CARD is commonly used for metagenomic ARG identification. The downstream analyses following identification are similar to those for other functional genes and can encompass a variety of approaches. It is essential to closely link these analyses to experimental design to ensure the accuracy and relevance of the results.
References
- B. P. Alcock, A. R. Raphenya, T. T. Y. Lau, K. K. Tsang, M. Bouchard, A. Edalatmand, W. Huynh, A.-L. V. Nguyen, A. A. Cheng, S. Liu, S. Y. Min, A. Miroshnichenko, H.-K. Tran, R. E. Werfalli, J. A. Nasir, M. Oloni, D. J. Speicher, A. Florescu, B. Singh, M. Faltyn, A. Hernandez-Koutoucheva, A. N. Sharma, E. Bordeleau, A. C. Pawlowski, H. L. Zubyk, D. Dooley, E. Griffiths, F. Maguire, G. L. Winsor, R. G. Beiko, F. S. L. Brinkman, W. W. L. Hsiao, G. V. Domselaar, A. G. McArthur, CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Research. 48, D517–D525 (2020).
- A. Cassini, L. D. Högberg, D. Plachouras, A. Quattrocchi, A. Hoxha, G. S. Simonsen, M. Colomb-Cotinat, M. E. Kretzschmar, B. Devleesschauwer, M. Cecchini, D. A. Ouakrim, T. C. Oliveira, M. J. Struelens, C. Suetens, D. L. Monnet, Burden of AMR Collaborative Group, Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. The Lancet. Infectious Diseases. 19, 56–66 (2019).
- J. Wang, J. Gu, X. Wang, Z. Song, X. Dai, H. Guo, J. Yu, W. Zhao, L. Lei, Enhanced removal of antibiotic resistance genes and mobile genetic elements during swine manure composting inoculated with mature compost. Journal of Hazardous Materials. 411, 125135 (2021).