
Methyl-Seq, a methylation target capture platform, provides a highly reliable method with single-base resolution for studying methylated genomic regions. The cataloged products offer methyl-seq designs for human, mouse, and rat. This platform offers unique advantages compared to common methylation detection platforms. By detecting individual CpGs, it provides more information than methylation arrays and offers higher throughput and cost-effectiveness than whole-genome bisulfite sequencing. It enables the discovery of methylated regions that cannot be detected by restriction enzyme-based approaches or immunoprecipitation methods.
Methyl-Seq Technical Advantages
Mouse: CpG design for approximately 100 Mb region; Rat: CpG design for approximately 90 Mb region.
High sensitivity: Single-base resolution.
Advanced bioinformatics analysis platform.
Probe-independent of methylation status.
High sensitivity: Resolution down to the single-base level.
Cost-effective: Increased throughput and reduced costs compared to whole-genome bisulfite sequencing.
Reduced bias compared to existing methylation methods.
Methyl-Seq Workflow
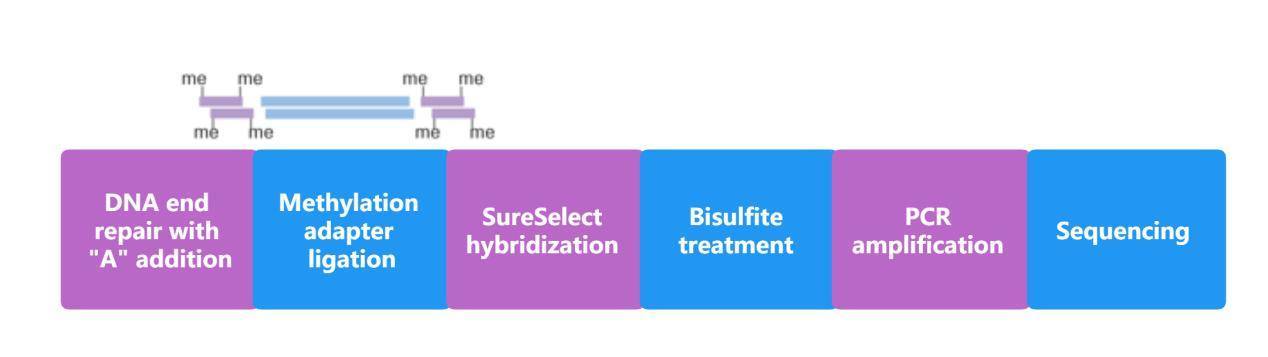 Optimized workflow of Methyl-Seq system for DNA methylation analysis using Agilent SureSelect target enrichment system.
Optimized workflow of Methyl-Seq system for DNA methylation analysis using Agilent SureSelect target enrichment system.
Methyl-Seq Data Analysis
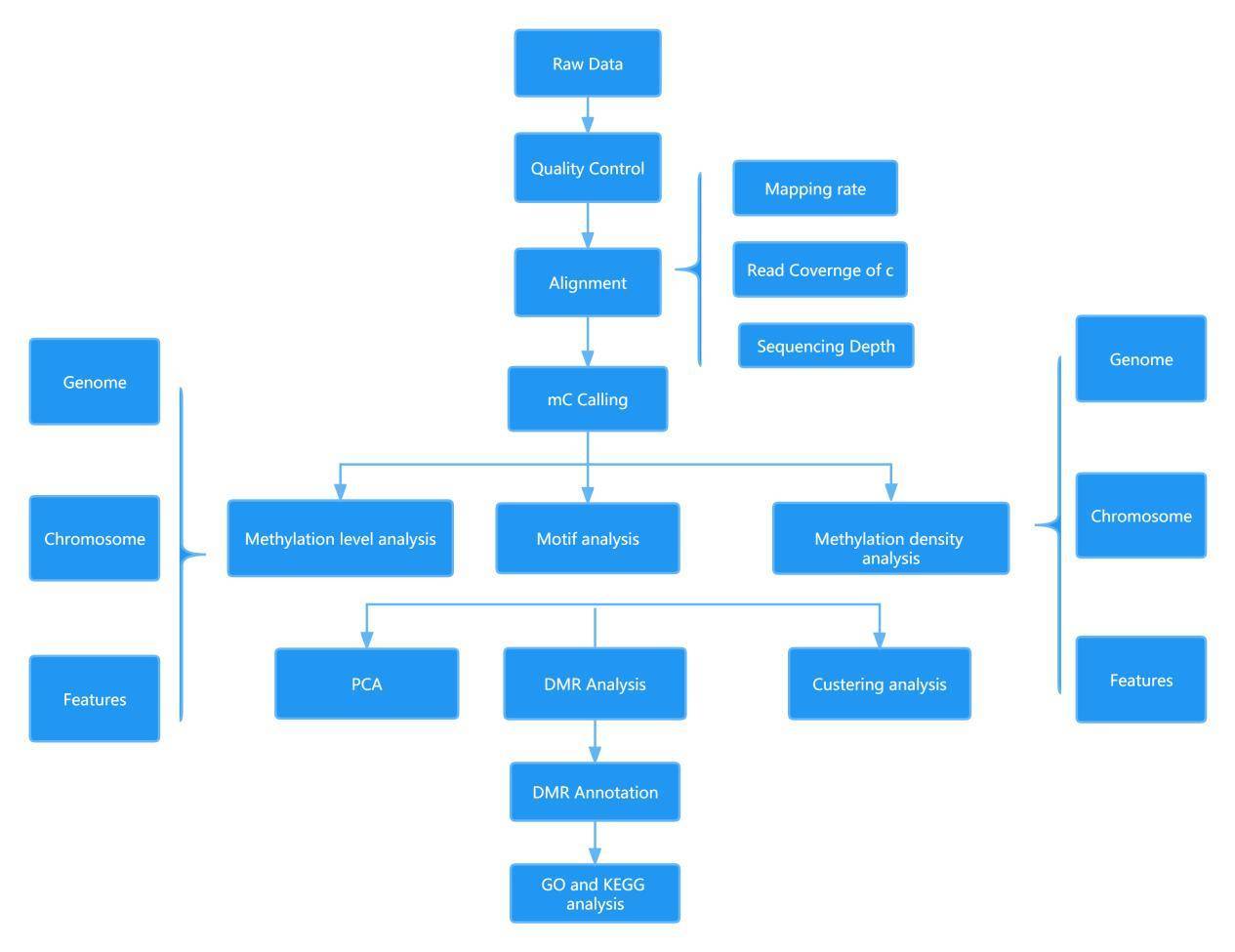
The DNA methylation analysis using the Epitranscriptomics Cloud Analysis tool developed by CD Genomics allows for simple and rapid visualization of methylation levels, CpG region coverage, and base quality. By identifying methylation states at single-base resolution, it enables the discovery of differentially methylated regions (DMRs) and facilitates the association analysis between methylation and transcription, WES, and other data. It also enables the identification and biological functional exploration of important methylation regions.
Methyl-Seq Application
Widely applied in drug models of mice and rats, stem cell research, tumor studies, and other complex disease investigations, it is particularly suitable for the discovery of epigenomic biomarkers and mechanism exploration in the whole epigenome of mice and rats.
Sample Requirements Sample Types
Human, mouse, and rat Sample Purity: OD 260/280 ratio should be between 1.8 and 2.0; RNA should be thoroughly removed Sample Concentration: DNA concentration should be accurately quantified using Qubit Sample Total Amount: The initial total DNA amount for each sample should be no less than 1 μg.
Methylation Research in Embryo Models
2i, an inhibitor of Mek1/2 and Gsk3β, LIF, a leukemia inhibitory factor, and S/L, a combination of serum and leukemia inhibitory factor, can induce mouse embryonic stem (ES) cells to a state similar to the pre-implantation embryo inner cell mass (ICM) with short-term treatment of 2i/L. Prolonged exposure to 2i/L leads to a series of irreversible epigenetic and genomic changes in mouse ES cells, impairing their developmental potential, which is caused by the long-term inhibition of Mek1/2.
The results indicate that derivatives of 2i/L ES cells exhibit a widespread decrease in DNA methylation, including methylation derived from gametes. These embryonic stem cells show damage during embryo and placental development, highlighting the limitations of 2i/L-established embryonic stem cells in terms of pluripotent cell quality. The authors' findings are of significant importance for understanding the implications of human naive pluripotent stem cells in therapeutic applications and disease modeling, as these cells often lose imprints and maternal methylation memory. The authors also discovered that early passage female a2i/L ES cells and t2i/L ES cells retain gametic DNA methylation and autonomous developmental potential. They propose that these embryonic stem cells can serve as an alternative cell culture platform for studying early embryonic development.
Reference
- Yagi M, Kishigami S, Tanaka A, et al. Derivation of ground-state female ES cells maintaining gamete-derived DNA methylation.Nature, 2017, 548(7666): 224.