





We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
The total length of DNA within human cells approximates two meters and necessitates complex folding processes to be accommodated within the nucleus. During this dynamic folding process, chromatin exhibits heterogeneous structural states, manifesting as either open or closed configurations. The open state of chromatin is crucial for the regulation of gene expression: transcription predominantly occurs in regions where chromatin is more accessible. Additionally, DNA methylation, histone modifications, and the binding of transcription factors require access to these open chromatin regions.
Open chromatin, characterized by a lower degree of compaction, provides a platform for the action of transcriptional regulatory proteins, forming the foundation for gene transcription and epigenetic regulation. The degree of chromatin folding and the presence of open regions are focal points of epigenomic research. Studies on chromatin accessibility can elucidate the molecular mechanisms of gene regulation and epigenetic changes, revealing the interaction networks between various transcriptional regulatory factors and downstream gene expression from a structural perspective.
In eukaryotic cells, chromatin serves as the fundamental unit of heredity, composed of DNA and histone proteins, and regulates cell-specific gene expression (Jackson Dean A., 1995; Bártová Eva and Kozubek Stanislav, 2012). As a dynamic nuclear structure, chromatin exhibits transcriptional activity during interphase and relative inactivity during metaphase of the cell cycle (Pederson Thoru, 2004). Transcriptional regulation involves the dynamic interactions between chromatin structure and numerous transcription factors recruited to enhancers, upstream activator sequences, and proximal promoter elements. These transcription factors facilitate the recruitment of RNA polymerase to core promoters, initiating transcription (Gottesfeld Joel M. and Carey Michael F., 2018).
Generally, regulatory elements selectively localize to accessible chromatin, which is essential for transcriptional regulation (Thurman et al., 2012). Although the occupancy of transcription factors is not always positively correlated with chromatin accessibility (Hsiung et al., 2015), maintaining an accessible chromatin conformation is necessary for the activation of target genes through transcription factor binding (Morris et al., 2014). Conversely, heterochromatin (or closed chromatin) restricts the binding of transcription factors and regulatory elements to promoters or enhancers, leading to gene silencing (Stergachis et al., 2013; Rinn J L and Chang H Y., 2012; Chen T and Dent S Y R, 2014). In summary, gene transcription necessitates the unwinding of higher-order DNA structures, though not entirely; only the regions of genes to be expressed need to be opened. This process is primarily facilitated by chromatin histone modifications, particularly acetylation. These partially opened chromatin regions are referred to as open chromatin, and once opened, they permit the binding of regulatory proteins, such as transcription factors.
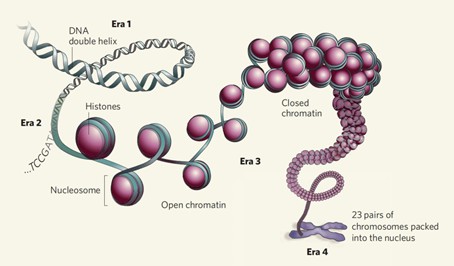 Figure 1 Open Chromatin vs Closed Chromatin
Figure 1 Open Chromatin vs Closed Chromatin
Chromatin accessibility refers to the degree of openness of chromatin, primarily facilitated by histone modifications. This property indicates the extent to which regulatory factors can bind to open chromatin regions and is closely associated with transcriptional regulation. Investigating regions of open chromatin in specific cellular states provides insights into transcriptional regulatory mechanisms at the DNA level.
Research in epigenetics predominantly employs high-throughput, genome-wide methodologies to study chromatin accessibility. Traditional experimental approaches, including Micrococcal MNase-seq and DNase-seq, operate on the principle that chromatin becomes more accessible when its structure is relaxed, resulting in decreased aggregation of DNA and histones. This exposure of DNA allows it to be cleaved by nucleases such as MNase or DNase I. Sequencing of the cleaved DNA fragments, followed by comparison with known genomic sequences, enables the identification of regions with increased accessibility. However, these methods are often labor-intensive and exhibit low reproducibility.
Another technique, FAIRE-seq, involves ultrasonic lysis followed by phenol-chloroform extraction, circumventing the need for nucleases or antibodies. Despite this advantage, FAIRE-seq suffers from high background noise, low signal-to-noise ratios, and challenges in optimizing formaldehyde crosslinking times.
ChIP-seq is employed to identify the binding sites of specific transcription factors or protein complexes, thereby elucidating DNA-protein interactions. This method utilizes antibodies to enrich for protein-DNA complexes, followed by sequencing of the associated DNA.
Although these techniques share a common analytical framework of identifying enriched regions and performing functional analyses, each has distinct limitations. In comparison, Assay for ATAC-seq utilizes the Tn5 transposase and is advantageous due to its simplicity, high reproducibility, and minimal requirement for cell or tissue samples. Additionally, ATAC-seq generates highly robust signals, making it the preferred technique for studying chromatin accessibility.
Service you may intersted in
Learn more
ATAC-seq, or Assay for Transposase-Accessible Chromatin with high-throughput sequencing, is a method developed in 2013 by the laboratories of William J. Greenleaf and Howard Y. Chang at Stanford University for investigating chromatin accessibility (often referred to as chromatin openness).
ATAC-seq represents an innovative technique in epigenetic research, leveraging the highly active Tn5 transposase as a probe to cleave DNA sequences and thereby locate accessible regions of chromatin across the entire genome (Buenrostro et al., 2013). DNA transposition involves the relocation of DNA transposons from one region of the chromosome to another with the assistance of transposase enzymes (Chuong et al., 2017). For ATAC-seq, this process necessitates that the insertion sites be in open chromatin regions. The methodology involves the artificial introduction of transposases carrying known DNA sequence tags into the cell nucleus, where they cleave the open chromatin, yielding DNA fragments tagged with primer sequences. These tagged fragments are then amplified using the known sequence primers to construct sequencing libraries (Buenrostro et al., 2013).
The most commonly used transposase in this context is the Tn5 transposase, which preferentially inserts into accessible chromatin regions over inaccessible ones. Functioning as a probe, the Tn5 transposase employs a "cut-and-paste" mechanism to detect chromatin accessibility on a genome-wide scale. The resulting DNA fragments are marked with sequencing tags to identify unprotected regions of the DNA (Reznikoff William S., 1993; Haniford D.B. and Ellis M.J., 2015).
In simpler terms, the Tn5 transposase can randomly bind to and cleave DNA in open chromatin regions while simultaneously inserting adaptor sequences at the cleavage sites. By incubating the transposase complex, which includes red and green sequence tags, with the cell nuclei and subsequently performing PCR amplification using the known sequence tags, a library is formed. Sequencing this library reveals information about the open chromatin regions.
The construction of an ATAC-seq library involves three main steps: nuclei preparation, transposition, and amplification. Initially, the tissue or cells to be examined are suspended to form a homogeneous single-cell suspension, followed by incubation in a lysis buffer to generate crude nuclei (Figure 3A). Subsequently, the suspended nuclei are incubated in a transposition reaction mixture, producing DNA fragments (Figure 3B). Finally, the transposed DNA is amplified to generate a sequencing library (Figure 3C). The reaction between the transposase and the sample chromatin is a critical step in the ATAC-seq experiment (Picelli et al., 2014).
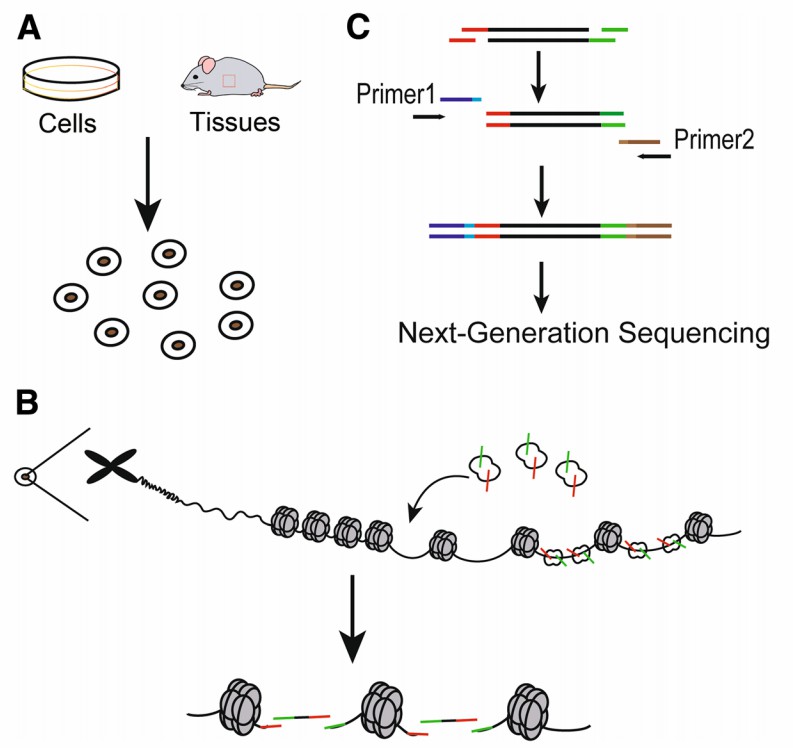 Figure 2 illustrates the main steps of ATAC-seq (Sun et al., 2019). (A) Nuclei Preparation: Target cells are lysed in lysis buffer to collect nuclei. (B) Transposase Reaction: Genomic DNA is tagged with Tn5 transposase. The green symbols represent "sequencing adapter 1" of the Tn5 transposase, while the red symbols represent "sequencing adapter 2". (C) PCR Amplification: Sequencing libraries are generated using PCR primers 1 and 2. Primers 1 and 2 are two universal PCR primers that capture specific length fragments and add barcodes suitable for next-generation sequencing.
Figure 2 illustrates the main steps of ATAC-seq (Sun et al., 2019). (A) Nuclei Preparation: Target cells are lysed in lysis buffer to collect nuclei. (B) Transposase Reaction: Genomic DNA is tagged with Tn5 transposase. The green symbols represent "sequencing adapter 1" of the Tn5 transposase, while the red symbols represent "sequencing adapter 2". (C) PCR Amplification: Sequencing libraries are generated using PCR primers 1 and 2. Primers 1 and 2 are two universal PCR primers that capture specific length fragments and add barcodes suitable for next-generation sequencing.
Note: Although the experimental process is illustrated using animal experiments, it is equally applicable to plants.
Prior to sequencing, ATAC-seq libraries require rigorous quality control to ensure that the library concentration meets sequencing standards. Sequencing is performed on libraries that meet these criteria, and raw reads are collected. Quality assessment and data filtering are subsequently conducted to obtain clean reads (Davie et al., 2015; Bell et al., 2011; Mardis, 2008). Post-filtering, high-quality reads approximately 150 nucleotides (nts) in length are processed for further analysis (Miskimen et al., 2017).
Peak calling involves mapping reads to the reference genome to identify accessible chromatin regions, such as promoters, enhancers, and insulators (Kumasaka et al., 2016; Ackermann et al., 2016; Quillien et al., 2017). This allows for a series of detailed analyses, including determining the genome-wide distribution of reads, assessing peak length distribution, performing functional analysis of genes associated with identified peaks, examining the distribution of peaks within gene functional elements, and analyzing differential peaks between samples (Pranzatelli et al., 2018; Mu et al., 2012).
ATAC-seq, or Assay for Transposase-Accessible Chromatin with high-throughput sequencing, was initially developed as an alternative or complementary method to MNase-seq, FAIRE-seq, and DNase-seq (Table 1).
In MNase-seq and DNase-seq, the reduction in DNA and histone aggregation leads to the exposure of unprotected DNA, which can then be cleaved by nucleases such as MNase and DNase. By sequencing these cleaved DNA fragments and comparing them to a reference genome, regions of accessible chromatin can be identified. However, these methods are often time-consuming and exhibit poor reproducibility. FAIRE-seq involves the fixation of DNA with formaldehyde, followed by phenol-chloroform extraction to isolate exposed DNA. Despite its utility, FAIRE-seq suffers from high background noise, low signal-to-noise ratio, and challenges in optimizing formaldehyde crosslinking times.
ATAC-seq addresses several of these limitations by employing the Tn5 transposase to insert sequencing adapters into open chromatin regions, thereby allowing for the efficient identification of accessible chromatin with minimal starting material. This method is advantageous due to its simplicity, rapid execution, and high reproducibility, making it a preferred technique for chromatin accessibility studies.
Table 1 Comparison of several sequencing methods (Sun et al., 2019).
| Method | MNase-seq | DNase-seq | FAIRE-seq | ATAC-seq |
| Cell State | Any | Any | Any | Fresh cells or slowly cooled frozen cells |
| Principle | MNase digestion of DNA protected by proteins or chromatin-bound nucleosomes. | Preferential cleavage of DNA sequences lacking nucleosomes by DNaseI. | Separation of naked DNA based on formaldehyde fixation and phenol-chloroform extraction. | Insertion of Tn5 transposase into DNA sequences not protected by proteins or nucleosomes and excising them. |
| Target Region | Focuses on nucleosome positioning. | Accessible chromatin regions, concentrated at transcription factor binding sites. | Accessible chromatin regions. | Genome-wide accessible chromatin regions, including transcription factors and histone modifications. |
| Specific Features | 1. Large number of cells as starting material; 2. Accurate enzyme quantities required; 3. Localization of entire nucleosomes and inactive regulatory regions; 4. Detection of inactive regions by degrading active ones; 5. Standard analysis requires 150-200M reads. | 1. Large number of cells as starting material; 2. Complex sample preparation process; 3. Accurate enzyme quantities required; 4. Standard analysis requires 20-50M reads. | 1. Low signal-to-noise ratio complicates data analysis; 2. Results heavily reliant on formaldehyde fixation; 3. Standard analysis requires 20-50M reads. | 1. Less starting material required; 2. Standard analysis requires 20-50M reads by reducing sequencing depth; 3. Convenient access to genome-wide accessible chromatin regions; 4. Impact of mitochondrial data on result accuracy. |
Efficiency and Time Reduction: The utilization of a transposase-based approach markedly reduces the experimental duration to approximately 2-3 hours. This expedited timeline is achieved through a direct enzymatic reaction for DNA fragmentation, obviating the labor-intensive conventional procedures of DNA shearing, end-repair, and adapter ligation.
Simplified Protocol: The streamlined experimental workflow minimizes sample preparation duration and diminishes the likelihood of errors, consequently enhancing the success rate and reproducibility of experiments. In contrast, DNase-seq and MNase-seq protocols typically entail 2-3 days, while FAIRE-seq necessitates 3-4 days.
Reduced Sample Requirement: The prerequisite sample input is substantially reduced by at least 1000-fold, from 1 million cells (FAIRE-seq) and 50 million cells (DNase-seq) to approximately 500 cells. This reduction is particularly advantageous in scenarios where sample collection poses challenges.
High-Resolution Mapping: ATAC-seq leverages paired-end sequencing to generate intricate nucleosome positioning and occupancy maps. Paired-end sequencing enables the sequencing of both ends of DNA fragments, thereby facilitating precise alignment of reads to repetitive genomic regions.
Enhanced Reproducibility and Simplicity: The reproducibility of ATAC-seq surpasses that of MNase-seq and DNase-seq. Moreover, the methodology is characterized by its simplicity of execution, minimal sample input requirements, and production of superior-quality sequencing signals.
Single-Cell Sequencing Capability: Recent advancements underscore the significance of single-cell sequencing in personalized epigenetic investigations. Conventional techniques such as ChIP-seq, DNase-seq, and MNase-seq are incompatible with single-cell analyses. However, ATAC-seq has been experimentally validated for single-cell sequencing, rendering it a cornerstone in contemporary epigenetic research.
Random Sequencing Adapters: The Tn5 transposase employs a "cut-and-paste" mechanism, fragmenting and tagging DNA in unprotected regions with sequencing adapters. Each DNA fragment's sequencing adapters at both ends are random, resulting in a 50% chance of both ends having identical sequencing adapters. Consequently, approximately half of the generated fragments are unusable for subsequent enrichment, amplification, and sequencing.
Preference for Unbound DNA and Transcription Factor Binding Sites: Studies indicate that "naked" DNA devoid of nucleosomes and transcription factors is more susceptible to cleavage by Tn5 transposase. Additionally, Tn5 transposase tends to bind and cleave in transcription factor binding regions, leading to partial loss of transcription factor information. These limitations render ATAC-seq challenging for detecting transcription factor footprints, which are crucial for identifying potential binding motifs.
Inclusion of Mitochondrial Reads: Due to the presence of mitochondrial DNA, data obtained through ATAC-seq inevitably contain some mitochondrial reads. Depending on the cell type, ATAC-seq data may comprise 20-80% mitochondrial sequencing reads.
ChIP-seq and assay for transposase-accessible chromatin using sequencing (ATAC-seq) are distinct methodologies employed in the study of chromatin dynamics and transcription factor binding.
ChIP-seq: This technique is predicated on the pre-identification of a specific transcription factor of interest. An antibody targeting the specific transcription factor is utilized to precipitate the DNA bound to it during the ChIP process. This enables the subsequent sequencing of the co-precipitated DNA, thereby validating the interaction between the transcription factor and the DNA.
ATAC-seq: In contrast, ATAC-seq does not focus on any specific transcription factor. Instead, it assesses the overall chromatin accessibility across the entire genome. This technique identifies potential binding sites for various proteins based on the openness of chromatin regions. By combining ATAC-seq with other methodologies, researchers can screen for and identify regulatory elements and factors of interest on a genome-wide scale.
In summary, while ChIP-seq provides insights into the binding interactions of specific transcription factors with DNA, ATAC-seq offers a broader perspective on chromatin accessibility and potential protein binding sites across the genome.
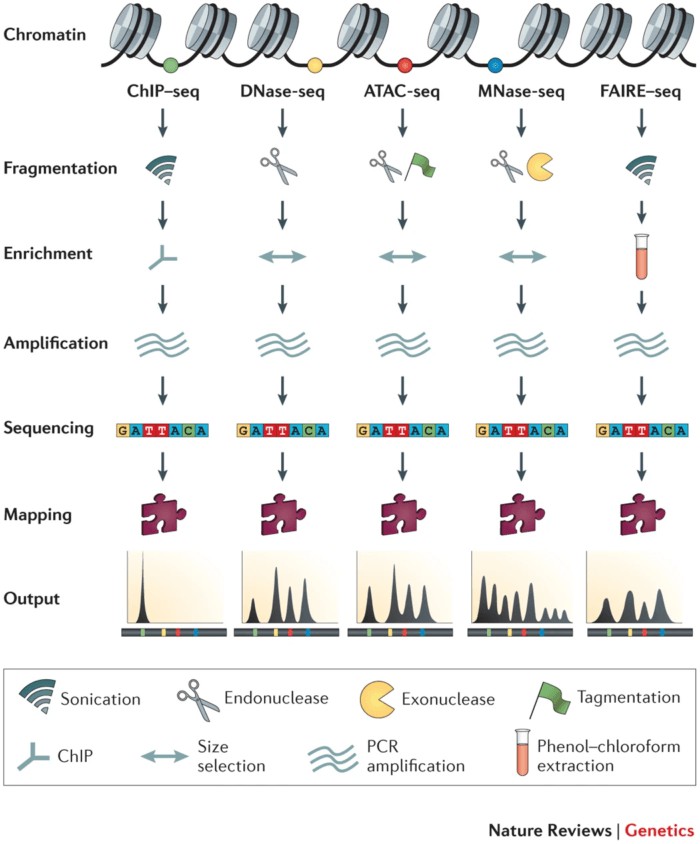 Figure 3 High-Throughput Methodologies Studying Chromatin Accessibility
Figure 3 High-Throughput Methodologies Studying Chromatin Accessibility
The Assay for ATAC-seq is extensively employed for the delineation of nucleosome positions across the genome, thereby providing significant insights into chromatin organization. Buenrostro et al. (2013) demonstrated the application of ATAC-seq in profiling nucleosome occupancy and chromatin accessibility in human cell lines, elucidating patterns of nucleosome positioning associated with regulatory elements. This study underscored the efficacy of ATAC-seq in generating high-resolution nucleosome positioning maps, which are essential for understanding the regulatory landscape of the genome.
The analysis of open chromatin regions via Assay for ATAC-seq facilitates the identification of transcription factors implicated in gene regulation. Corces et al. (2016) utilized ATAC-seq to profile chromatin accessibility across diverse human tissues, identifying transcription factors such as CTCF (CCCTC-binding factor) and AP-1 (Activator Protein 1) as crucial regulators of gene expression. This investigation highlighted the importance of ATAC-seq in elucidating transcription factors that play essential roles in cell-specific regulatory networks.
ATAC-seq is adept at identifying open chromatin regions that correspond to promoters and distal regulatory elements, encompassing enhancers and silencers. Thurman et al. (2012) employed ATAC-seq to map DNase I hypersensitive sites within the human genome, thereby revealing active regulatory elements such as promoters and enhancers. This methodology is instrumental in elucidating the regulatory elements that drive gene expression and in understanding the complex interactions within the genome.
The integrative approach of combining Assay for ATAC-seq with other omics technologies, such as RNA sequencing, provides a comprehensive perspective on gene regulation. For instance, Satpathy et al. (2019) employed a combination of ATAC-seq and single-cell RNA-seq to investigate the immune cell landscape in melanoma, thereby identifying regulatory elements associated with gene expression changes across different cell states. This integrative methodology enhances the understanding of how chromatin accessibility influences gene expression and cellular function.
Assay for ATAC-seq generates comprehensive genome-wide maps of chromatin accessibility, imperative for investigating dynamic changes in chromatin structure. Schep et al. (2015) utilized ATAC-seq to map chromatin accessibility during mouse embryonic stem cell differentiation, revealing dynamic alterations correlating with gene expression patterns. These maps are crucial for elucidating the epigenetic regulation of developmental processes.
Regulated by Transcription Factors, Assay for ATAC-seq aids in identifying target genes controlled by transcription factors by locating regions of open chromatin where these factors bind. Heinz et al. (2018) employed ATAC-seq to pinpoint binding sites of transcription factors such as PU.1 and CEBPA in macrophages, elucidating their roles in regulating immune response genes. This application is vital for mapping the regulatory networks that govern gene expression in diverse biological contexts.
ATAC-seq can be utilized for the characterization of transcription factor binding sites through footprinting analysis. Pique-Regi et al. (2011) applied ATAC-seq footprinting to identify protected regions within open chromatin, indicative of direct binding sites for transcription factors such as NF-κB (Nuclear Factor kappa-light-chain-enhancer of activated B cells) and p53. This technique provides detailed insights into the binding dynamics of transcription factors and elucidates their regulatory roles.
Title: ATAC-seq and RNA-seq reveal the role of AGL18 in regulating fruit ripening via ethylene-auxin crosstalk in papaya
Methods: ATAC-seq, RNA-seq, Subcellular Localization, Y1H, Dual Luciferase Reporter Gene Assay, ChIP-qPCR, EMSA
Abstract:
This study employs integrated ATAC-seq and RNA-seq to delineate chromatin accessibility patterns under different 1-methylcyclopropene (1-MCP) treatments, uncovering key transcription factors and target genes influencing fruit ripening. The approach is straightforward yet informative, conducive to producing publications of approximately five points.
Background:
Papaya (Carica papaya) stands as a quintessential tropical fruit, prized for its distinctive flavor and rich nutritional profile. However, papaya is a climacteric fruit, undergoing rapid physiological changes during ripening, rendering it highly susceptible to rot, thereby posing significant challenges for its transportation and storage (Sivakumar et al., 2013). Studies have shown that 1-methylcyclopropene (1-MCP), an ethylene receptor inhibitor, can delay ripening and rotting in fruits such as bananas, apples, and papayas, with appropriate 1-MCP treatment postponing papaya fruit yellowing and softening. Nonetheless, improper 1-MCP treatment can impede papaya ripening, resulting in fruits that fail to soften adequately and retain high hardness even at the end of storage, severely limiting the application of 1-MCP in postharvest papaya industries. Various researches indicate the pivotal roles of auxin and ethylene in fruit ripening. However, the molecular mechanisms underlying the interaction between auxin and ethylene during papaya fruit ripening remain elusive (Petrasek et al., 2009; Tian et al., 2006).
Researchers employed integrated ATAC-seq and RNA-seq analyses to unveil differences in chromatin accessibility under 1-MCP treatments of varying durations during papaya ripening, aiming to identify key differentially accessible regions (DARs), upstream motifs, and transcription factors (TFs) implicated in delaying and inducing abnormal papaya ripening under 1-MCP treatment. This elucidation aids in identifying genes that modulate the interplay between the ethylene and auxin signaling pathways associated with papaya ripening, thereby serving as candidate genes for improving fruit quality and postharvest shelf life (Cai et al., 2022).
Experimental Design:
Fruits were divided into three groups: short-term treatment with 400 nL/L 1-MCP for 2 hours, long-term treatment with 400 nL/L 1-MCP for 16 hours, and untreated control continuously kept in a sealed foam box for 16 hours. Samples were collected one and six days after treatment, with three biological replicates for RNA-seq and two biological replicates for ATAC-seq (Figure 4).
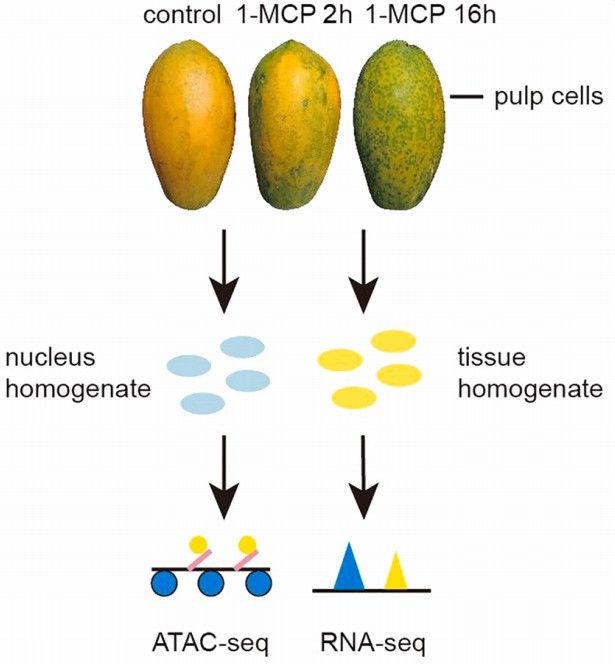 Figure 4 illustrates the workflow of ATAC-seq and RNA-seq on papaya fruits post 1-MCP treatment. Blue ellipses represent nuclear homogenate, while yellow ellipses represent tissue homogenate.
Figure 4 illustrates the workflow of ATAC-seq and RNA-seq on papaya fruits post 1-MCP treatment. Blue ellipses represent nuclear homogenate, while yellow ellipses represent tissue homogenate.
Research Workflow:
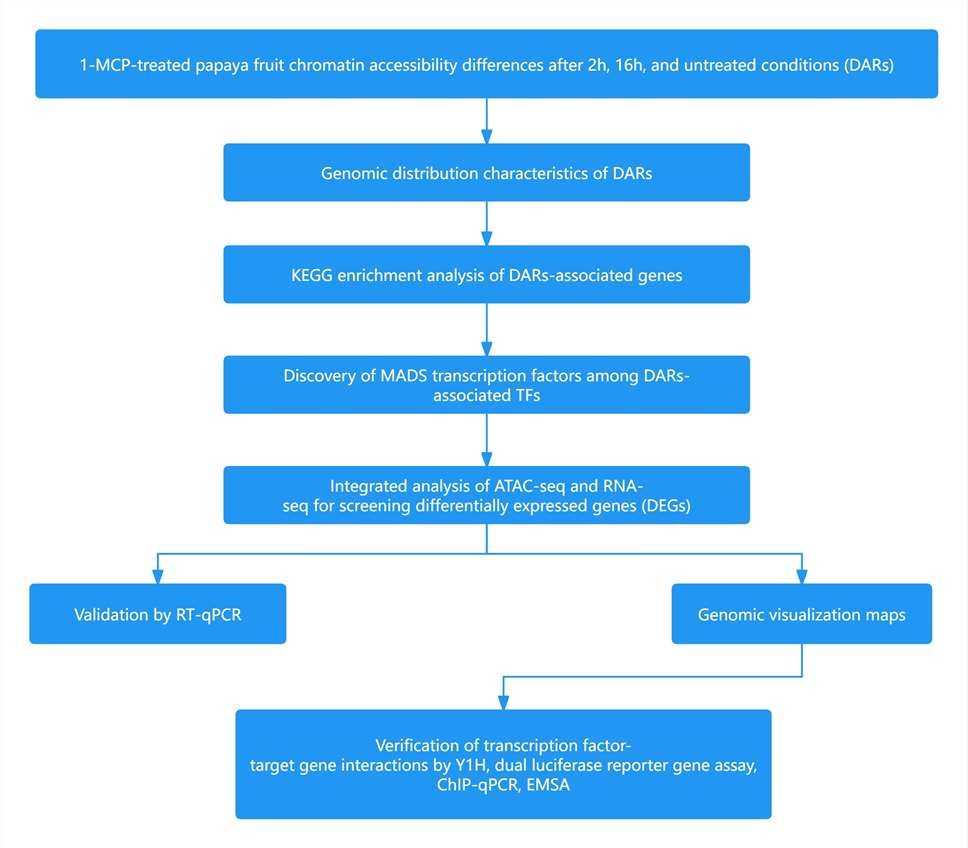 Figure 5 Research Strategy of Case 1
Figure 5 Research Strategy of Case 1
Research Results:
Chromatin Accessibility in Papaya Fruits under Various 1-MCP Treatments
The alterations in chromatin accessibility in papaya fruits subjected to short-term and long-term 1-MCP treatments were analyzed using Assay for Transposase-Accessible Chromatin with high-throughput sequencing (ATAC-seq). Principal component analysis (PCA) (Figure 6A) demonstrated high Pearson correlation coefficients among biological replicates: 0.96 for the control group, 0.98 for the 2-hour 1-MCP treatment group, and 0.98 for the 16-hour 1-MCP treatment group, indicating the reliability of the sampling and data.
Volcano plots of differentially accessible regions (DARs) (Figure 6B) revealed that, compared to the control group, 1179 and 1219 DARs were identified in the 2-hour and 16-hour 1-MCP treated samples, respectively. Furthermore, 592 DARs were identified when comparing the 16-hour treatment samples to the 2-hour treatment samples.
The chromatin accessibility in papaya samples treated with 1-MCP at different time points was predominantly located in intergenic regions and promoter regions, with a significant relationship with promoters. Notably, downregulated DARs in the 16-hour 1-MCP treated samples were significantly distributed in promoter regions compared to the control group. These findings suggest that 1-MCP treatment markedly alters chromatin accessibility (Figure 6C).
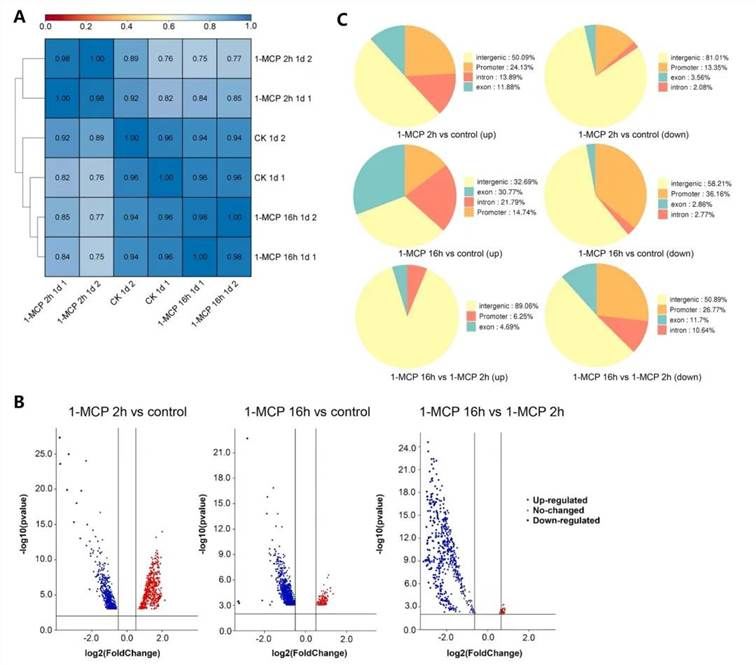 Figure 6 illustrates the differential chromatin accessibility in papaya fruits treated with 1-MCP for varying durations.
Figure 6 illustrates the differential chromatin accessibility in papaya fruits treated with 1-MCP for varying durations.
(A) Pearson Correlation Clustering Heatmap: This heatmap is based on ATAC-seq read counts, demonstrating high correlation coefficients among biological replicates, indicating the reliability of the data and sampling.
(B) Volcano Plot of Differentially Accessible Regions (DARs): The volcano plot presents DARs under different 1-MCP treatments. Significance is indicated by an FDR ≤ 0.05 and a fold change ≥ 1.2 or ≤ 0.8. Upregulated DARs are represented by red circles, while downregulated DARs are indicated by blue circles.
(C) Genomic Distribution of DARs: The distribution of DARs across genomic regions (promoters, introns, coding exons, and distal intergenic regions) in papaya fruit samples under various treatments is depicted.
KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment analysis of genes associated with upregulated or downregulated DARs due to 1-MCP treatment revealed significant involvement of plant hormone signal transduction pathways. These pathways are crucial in the inhibition of fruit ripening and softening induced by 1-MCP.
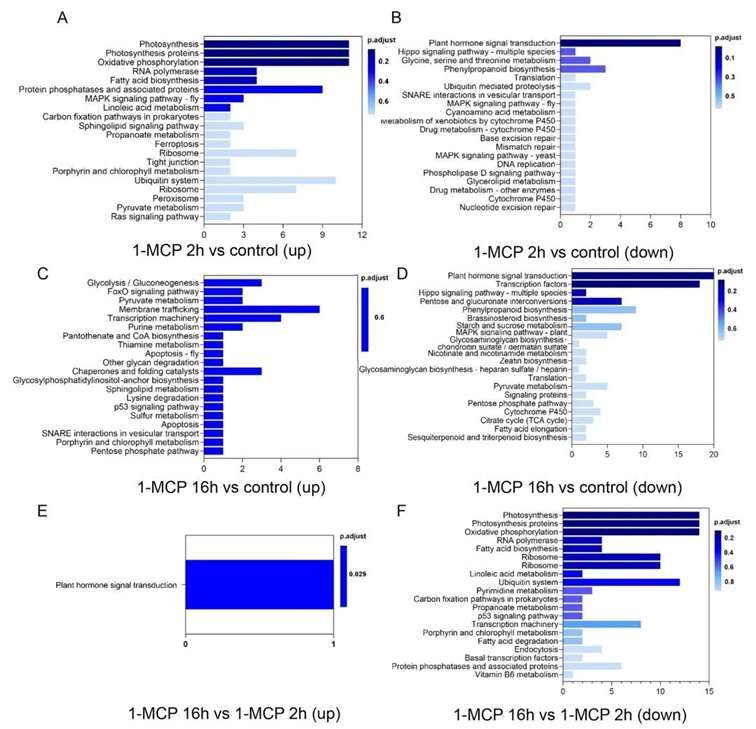 Figure 7 displays the Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment bar graph of genes associated with Differentially Accessible Regions (DARs) in papaya fruit under different treatments of 1-MCP.
Figure 7 displays the Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment bar graph of genes associated with Differentially Accessible Regions (DARs) in papaya fruit under different treatments of 1-MCP.
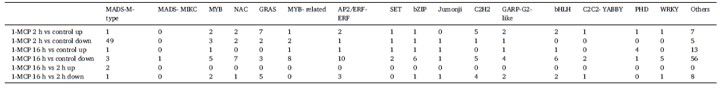
The analysis of transcription factors (TFs) associated with differentially accessible regions (DARs) revealed a predominant downregulation of TFs in samples treated with 1-MCP for both 2 hours and 16 hours, compared to the control. Among these, MADS-M type TFs constituted the majority of downregulated TFs in the 2-hour 1-MCP treatment group compared to the control. In contrast, MADS-M type TFs were the only differentially expressed TFs that were upregulated when comparing the 16-hour treatment to the 2-hour treatment.
Additionally, various other TFs, including MYB, NAC, and AP2/ERF-ERF, exhibited significant changes under different 1-MCP treatments, indicating their involvement in the regulation of papaya fruit ripening. These findings underscore the pivotal role of MADS-M type TFs, also known as MADS-box TFs, in the maturation process of papaya under 1-MCP treatment (Table 1).
Previous studies have demonstrated that MADS-box TFs regulate plant maturation through multiple mechanisms. For instance, in tomatoes, SlMBP9, a MADS-box gene, regulates PIN expression and reduces auxin transport, thus modulating apical dominance and lateral root formation, while SlAGL15 regulates ARF6/8 to participate in auxin-related signaling pathways (Li et al., 2019). In papaya, CpMADS1/3 regulates CpEIL1, thereby participating in ethylene signal transduction (Ding et al., 2019).
Table 1 illustrates the distribution of transcription factors (TFs) under various treatments of 1-MCP.
Integrated Analysis of ATAC-seq and RNA-seq Data
Using genome feature format (GFF) annotation for this species, the chromatin differentially accessible regions (DARs) under varying 1-MCP treatments were linked to their nearest associated genes. Short-term 1-MCP treatment identified 25 genes showing differential expression in both chromatin accessibility and transcriptomic analyses when compared to the control (Figure 8A). Long-term 1-MCP treatment revealed 187 genes with significant differences in both chromatin accessibility and transcriptomic profiles relative to the control (Figure 8B). Additionally, a comparison between long-term and short-term 1-MCP treatments identified 21 genes exhibiting differential expression in both chromatin accessibility and transcriptomic analyses (Figure 8C).
These genes, which display differences in both chromatin accessibility and transcriptomic analysis, likely play crucial roles in the ripening process of papaya fruit and in the inhibition of papaya fruit maturation by 1-MCP.
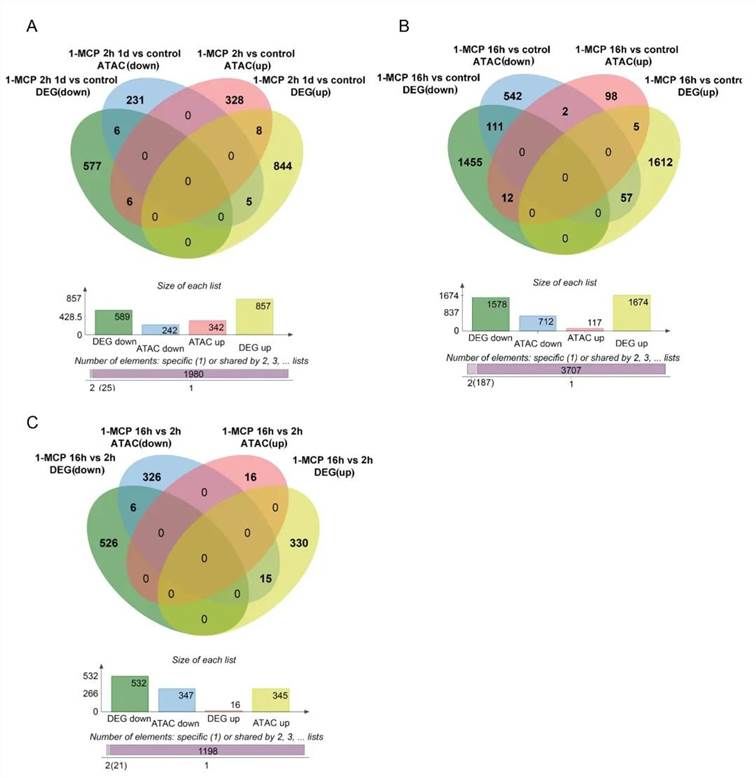 Figure 8 presents the differential analysis of chromatin accessibility and gene expression in papaya fruit samples under different treatments of 1-MCP. The Venn diagram illustrates the overlap between differentially expressed genes (DEGs) and genes associated with Differentially Accessible Regions (DARs).
Figure 8 presents the differential analysis of chromatin accessibility and gene expression in papaya fruit samples under different treatments of 1-MCP. The Venn diagram illustrates the overlap between differentially expressed genes (DEGs) and genes associated with Differentially Accessible Regions (DARs).
Differentially expressed genes (DEGs) were aligned to the genome using databases from NCBI (http://www.ncbi.nlm.nih.gov/) and JGI (https://phytozome.jgi.doe.gov/pz/portal.html). Genes associated with the ethylene signaling pathway (CpEIL3, CpERF113, CpACS1), auxin signaling pathway (CpWAT, CpSAUR32/50, CpPIN, CpTIR1), and CpAGL18 (a MADS-box gene) were selected for further validation using reverse transcription quantitative polymerase chain reaction (RT-qPCR).
In the control group, the expression levels of CpACS1, CpEIL3, CpERF113, CpSAUR32, CpSAUR50, CpTIR1, and CpAGL18 increased at 1 day after treatment (DAT) and subsequently decreased by 6 DAT (Figure 9). The application of 1-MCP significantly inhibited the expression levels of these genes, particularly in the long-term treatment group. Genes related to auxin, such as PIN and WAT, exhibited a similar expression pattern, with a decrease in expression during ripening, but an increase following long-term 1-MCP treatment. The expression profiles obtained via RT-qPCR were consistent with the RNA-seq results, underscoring the reliability of these expression data.
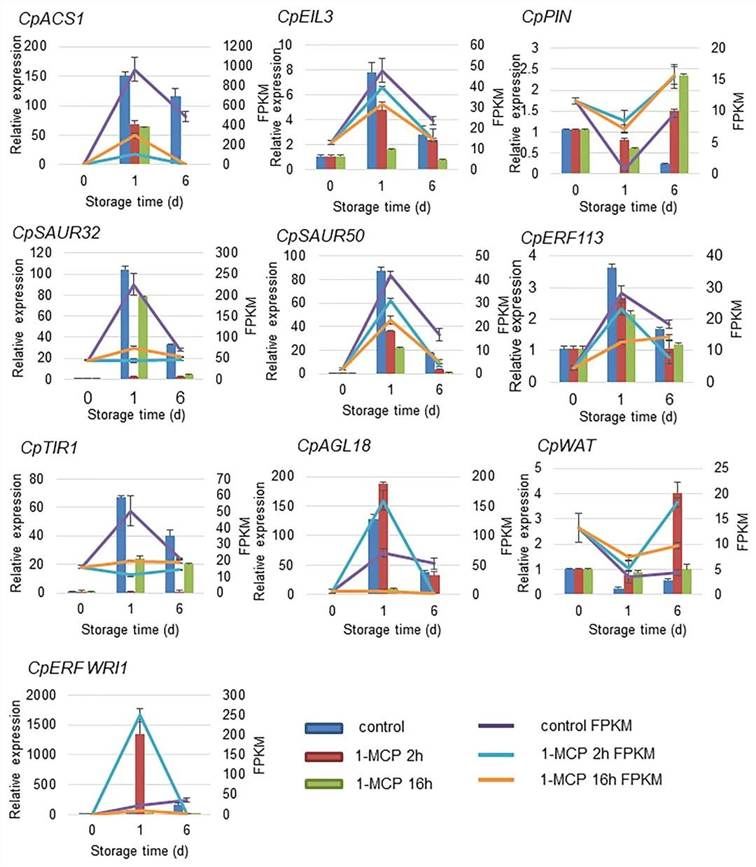 Figure 9 illustrates the expression validation of selected differentially expressed genes (DEGs) under various treatments of 1-MCP.
Figure 9 illustrates the expression validation of selected differentially expressed genes (DEGs) under various treatments of 1-MCP.
The integrated genomic visualization of ATAC-seq and RNA-seq data demonstrated differential chromatin accessibility and gene expression profiles in papaya fruit subjected to various 1-MCP treatments. Short-term 1-MCP treatment inhibited the expression of CpAGL18 and CpSAUR32 and reduced the associated chromatin openness compared to the control. Under long-term 1-MCP treatment, both CpACS1 and CpAGL18 exhibited decreased expression and chromatin accessibility relative to the control. Notably, CpAGL18 was consistently affected by both short-term and long-term 1-MCP treatments (Figure 10).
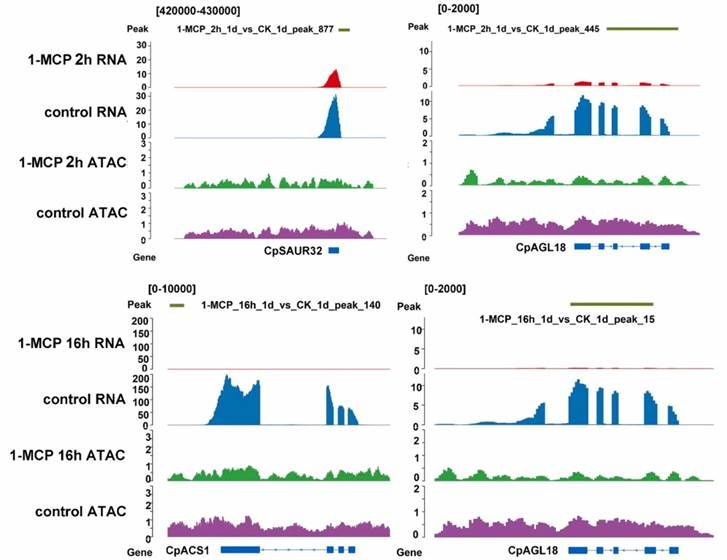 Figure 10 presents a genomic visualization map integrating ATAC-seq and RNA-seq data, illustrating the differential gene expression influenced by 1-MCP treatment and the chromatin accessibility changes associated with these genes.
Figure 10 presents a genomic visualization map integrating ATAC-seq and RNA-seq data, illustrating the differential gene expression influenced by 1-MCP treatment and the chromatin accessibility changes associated with these genes.
Experimental Validation of CpAGL18 Activation of CpACS1 and CpSAUR32 Transcription
The expression profile of CpAGL18 indicates its pivotal role in papaya fruit ripening. Within the key differentially expressed genes (DEGs) identified through the integrated RNA-seq and ATAC-seq analyses, CpACS1 and CpSAUR32 possess cis-regulatory elements for MADS-box binding sites. Subcellular localization analysis demonstrated that CpAGL18 is localized in both the nucleus and plasma membrane. Transient expression in Nicotiana benthamiana cells showed predominant fluorescence of CpAGL18 in the nucleus with a minor fraction at the plasma membrane, whereas the control GFP fluorescence was uniformly distributed throughout the cell (Figure 11A).
Yeast one-hybrid (Y1H) assays were conducted to evaluate the binding activity of the CpAGL18 protein to the promoter regions of CpACS1 and CpSAUR32. No promoter activity of CpACS1 and CpSAUR32 was observed in yeast cells without the recombinant CpAGL18. However, yeast cells transformed with the recombinant CpAGL18 activation domain (AD) and containing the CpACS1 and CpSAUR32 promoters exhibited robust growth on AbA selection medium, indicating that CpAGL18 binds to the promoters of CpACS1 and CpSAUR32 (Figure 11B). Furthermore, dual-luciferase reporter assays were employed to quantify the promoter activity of CpACS1 and CpSAUR32. The analysis revealed significantly higher LUC/REN ratios in the presence of CpAGL18 compared to the control, demonstrating that CpAGL18 markedly induces the expression of CpACS1 and CpSAUR32 (Figure 11C).
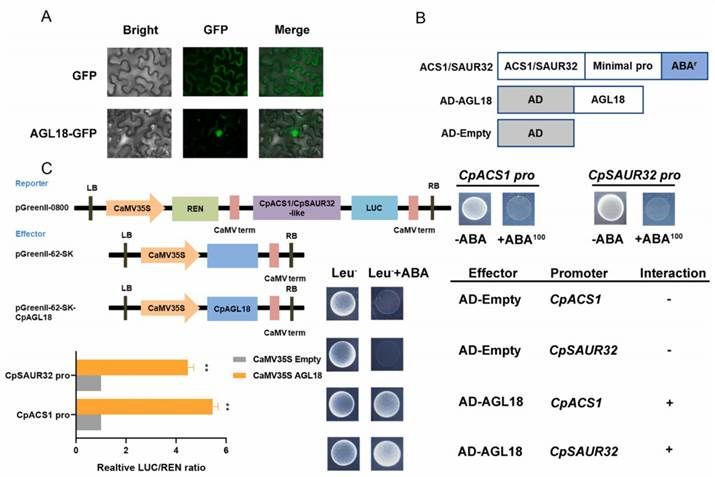 Figure 11 illustrates the detection of interactions between CpAGL18 and its target genes.
Figure 11 illustrates the detection of interactions between CpAGL18 and its target genes.
Electrophoretic mobility shift assay (EMSA) was employed to assess the direct interaction between CpAGL18 and the promoters of CpACS1 and CpSAUR32 in vitro. The results demonstrated that CpAGL18 binds to the CarG box elements within the CpACS1 and CpSAUR32 promoters, resulting in a noticeable shift in mobility. The inclusion of mutated promoter fragments did not significantly disrupt the formation of the DNA-protein complexes, thereby indicating the specificity of the interaction (Figure 12A).
Additionally, chromatin immunoprecipitation followed by quantitative PCR (ChIP-qPCR) analysis revealed a significant enrichment of CpAGL18 at the promoter regions of CpACS1 and CpSAUR32 when compared to the non-specific IgG antibody control (Figure 12B). These findings confirm the direct binding of CpAGL18 to the promoters of CpACS1 and CpSAUR32, further substantiating its role in regulating the transcription of these genes.
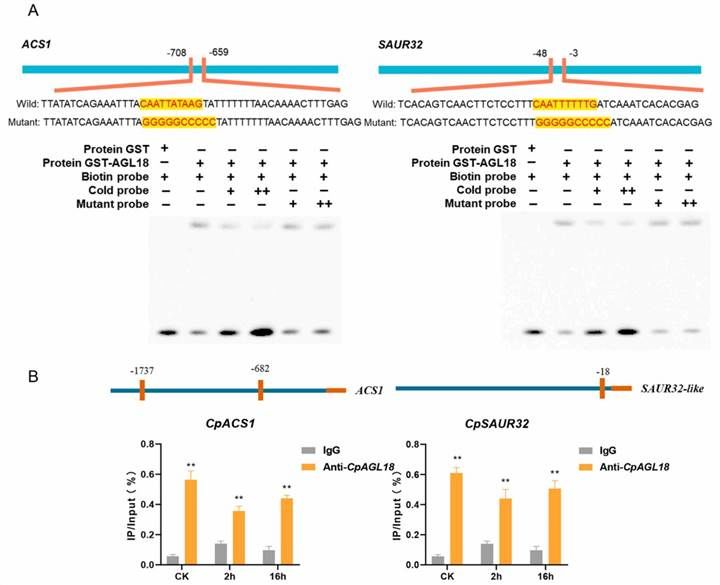 Figure 12 depicts the interaction between CpAGL18 and its target genes in vitro and in vivo.
Figure 12 depicts the interaction between CpAGL18 and its target genes in vitro and in vivo.
Conclusion:
Through ATAC-seq analysis, it has been observed that MADS transcription factors are involved in papaya fruit ripening.
The integrated analysis of ATAC-seq and RNA-seq reveals that CpAGL18, along with eight genes associated with auxin and ethylene, potentially play significant roles in fruit ripening.
Both in vivo and in vitro experiments demonstrate that CpAGL18 binds to and activates the expression of CpACS1 and CpSAUR32, thus participating in the ethylene and auxin signaling pathways, thereby influencing the regulation of papaya fruit ripening process.
Case 2
Title: A lamin-like protein OsNMCP1 modulates drought resistance and root growth by interacting with a chromatin remodeler OsSWI3C in rice
Technical Approaches: Subcellular localization, RT-qPCR, Western blotting (WB), ATAC-seq, RNA-seq, Yeast two-hybrid (Y2H), Co-immunoprecipitation (Co-IP), Bimolecular fluorescence complementation (BiFC)
The focus of this study revolves around the lamin-like protein OsNMCP1 and the chromatin remodeling complex OsSWI3C. Through a combined analysis of ATAC-seq and RNA-seq, the study investigates the chromatin accessibility and downstream gene expression under drought and normal conditions in OsNMCP1 overexpression transgenic lines and OsSWI3C interference transgenic lines, elucidating the roles of OsNMCP1 and OsSWI3C in regulating drought and root growth-related genes. While other experiments in this article are relatively comprehensive, the integration of ATAC-seq and RNA-seq analysis elevates the study to a higher level, making it particularly instructive for researchers aiming to publish high-quality articles.
Background: Nuclear envelope proteins in animals play crucial roles in nuclear functions, including chromatin organization, signal transduction, gene regulation, and cell differentiation. Nuclear Matrix Constituent Proteins (NMCPs) are lamin-like proteins in plants; however, their regulatory functions remain elusive (Stuurman et al., 1998; Dittmer et al., 2011). In preliminary experiments, researchers observed that drought stress induced the expression of a gene highly similar to NMCP1 (a putative lamin-like protein in plants). This gene was designated as OsNMCP1 (LOC_Os01g56140). Given the limited reports on lamin-like proteins in plants under drought conditions, the researchers aimed to investigate the role of this gene in drought resistance.
Research Strategy:
 Figure13 Research Strategy of Case 2
Figure13 Research Strategy of Case 2
Research Findings:
OsNMCP1 is a lamin-like nuclear envelope protein induced by drought stress.
Initially, subcellular localization experiments were conducted, confirming the peripheral nuclear localization of the lamin-like protein OsNMCP1 (Figure 14a-l). qRT-PCR analysis revealed an increase in the transcription levels of OsNMCP1 in both leaf and root tissues following treatment with Abscisic acid (ABA) and drought stress (Figure 14m-n). Additionally, Western blot (WB) analysis using specific antibodies against OsNMCP1 indicated an elevation in OsNMCP1 protein levels in leaf and root tissues under drought stress (Figure 14o). These findings suggest that OsNMCP1 responds to drought stress and ABA.
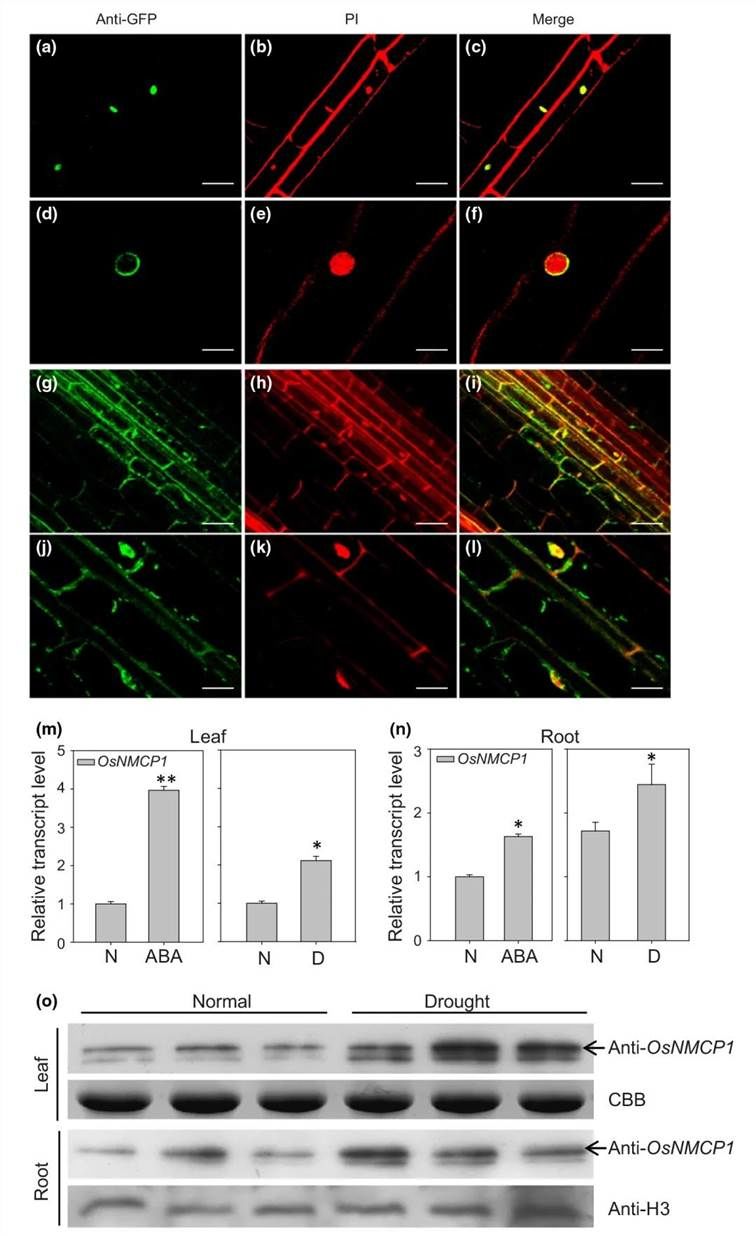 Figure 14 depicts the subcellular localization and expression profile of OsNMCP1.
Figure 14 depicts the subcellular localization and expression profile of OsNMCP1.
OsNMCP1 Positively Regulates Drought Resistance in Rice
To investigate the involvement of OsNMCP1 in regulating drought resistance in rice, two independent lines of OsNMCP1 overexpression (OE) transgenic rice (in the ZH11 background), OsNMCP1-OE-5 and OsNMCP1-OE-8, were generated, along with two allelic mutant lines harboring T-DNA insertions, osnmcp1-1 (PFG_3A-60922, in the DJ background) and osnmcp1-2 (PFG_1C-01360.R, in the HY background). OsNMCP1-OE lines exhibited higher survival rates post-drought stress recovery compared to ZH11 (Figure 15c, e). Conversely, the osnmcp1 mutants displayed increased sensitivity to drought stress (Figure 15d), with lower survival rates post-recovery compared to DJ/HY (Figure 15f). Consistent results were obtained across three independent replicates. These findings suggest that OsNMCP1 plays a promotive role in rice drought resistance.
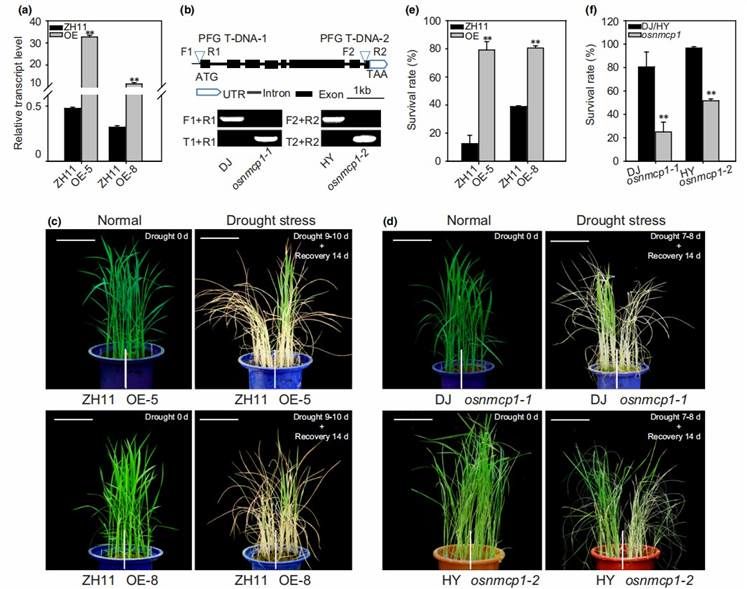 Figure 15. Phenotypic Analysis of Rice OsNMCP1 Mutants and OsNMCP1 Overexpressing (OE) Plants Under Drought Stress During the Seedling Stage
Figure 15. Phenotypic Analysis of Rice OsNMCP1 Mutants and OsNMCP1 Overexpressing (OE) Plants Under Drought Stress During the Seedling Stage
Due to the absence of significant differences in the wilting rates between wild-type and transgenic plant detached leaves, researchers assessed the maximum root length and root volume of plants at the seed maturation stage under both normal and drought stress conditions. Under normal growth conditions, OsNMCP1 overexpression (OE) lines exhibited longer root lengths and larger root volumes compared to ZH11, with more pronounced differences under drought stress conditions (Figure 16c, e, f). Conversely, the osnmcp1 mutants displayed noticeably shorter root lengths and smaller root volumes compared to DJ/HY under both normal and drought stress conditions (Figure 16d, g, h). These collective findings suggest that OsNMCP1 plays a positive role in regulating drought resistance, possibly attributed in part to enhanced root growth.
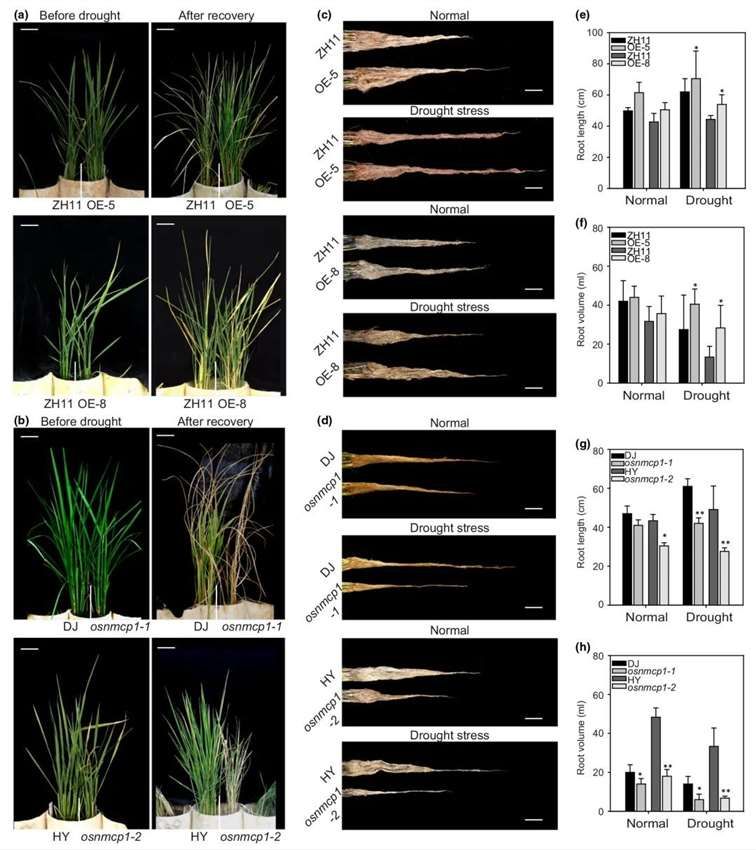 Figure 16 demonstrates the phenotypic responses of rice (Oryza sativa) lines, specifically OsNMCP1-overexpressing (OsNMCP1-OE) and osnmcp1 mutant lines, under drought stress conditions during the reproductive stage.
Figure 16 demonstrates the phenotypic responses of rice (Oryza sativa) lines, specifically OsNMCP1-overexpressing (OsNMCP1-OE) and osnmcp1 mutant lines, under drought stress conditions during the reproductive stage.
Regulation of Root Growth by OsNMCP1
Further investigation focused on the root phenotypes during the seedling stage. Compared to the corresponding wild-type (WT), OsNMCP1 overexpression (OE) lines exhibited elongated primary roots (Figure 17a), while osnmcp1 mutants displayed shortened primary roots (Figure 17b). The transcription levels of OsNMCP1 were analyzed via qRT-PCR across different root regions, including the meristematic (1-2 mm from root tip), elongation (2-6 mm), and mature (≥6 mm) zones, as well as the root cap (≤1 mm) (Figure 17c). The highest expression of OsNMCP1 was detected in the elongation zone, followed by the meristematic zone, with a decline observed in the mature zone and root cap (Figure 17d). To ascertain if cell division in roots of OsNMCP1-OE and osnmcp1 mutant lines was affected, 5-ethynyl-2'-deoxyuridine (EdU) staining was performed. A noticeable increase in EdU signal was observed in OsNMCP1-OE, whereas it decreased in osnmcp1 mutants compared to the WT (Figure 17e). These data suggest that OsNMCP1 may regulate root growth by modulating cell division. Given that auxin controls the size of the root meristem and the extent of cell division, the concentration of free indole-3-acetic acid (IAA) was measured in roots of OsNMCP1-OE-5 and osnmcp1-1. Results revealed an increase in IAA in OsNMCP1-OE-5 compared to the corresponding WT, while a decrease was observed in osnmcp1-1 mutants (Figure 17f).
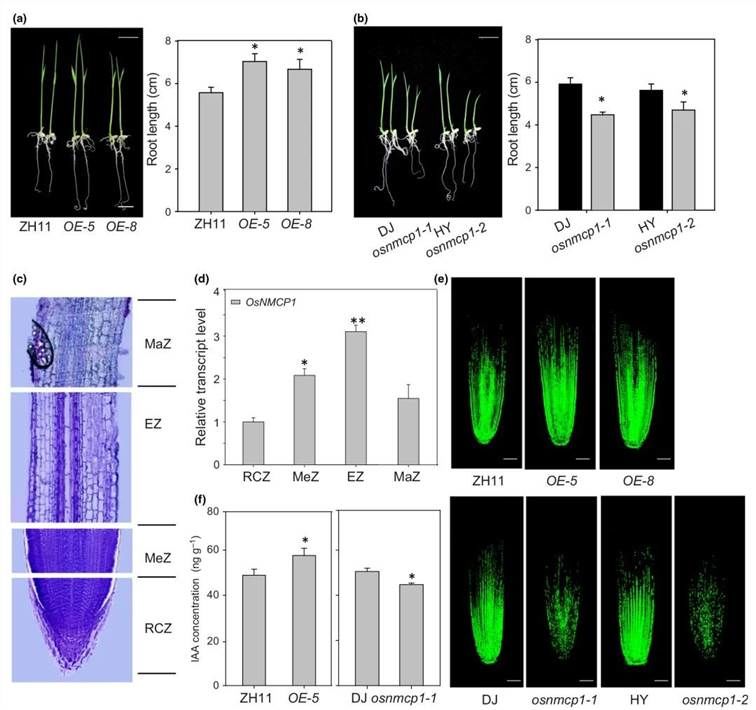 Figure 17 illustrates the influence of OsNMCP1 on the growth of rice roots and the rate of cell division. (a) Phenotypic characterization and statistics of primary root length in two independent lines of ZH11 and OsNMCP1-OE plants; (b) Phenotypic characterization and statistics of primary root length in Dongjing (DJ) and Huayoung (HY) varieties, as well as osnmcp1-1 and osnmcp1-2 mutant lines; (c) Morphology of the root tip including MaZ (mature zone), EZ (elongation zone), MeZ (meristematic zone), and RCZ (root cap zone); (d) Expression levels of OsNMCP1 in different root tip zones analyzed by qRT-PCR; (e) Longitudinal view of Edu-labeled cells in the root meristematic tissue of 7-day-old seedlings of OsNMCP1-OE, osnmcp1 mutants, and corresponding wild-type (WT) plants; (f) Statistics of endogenous auxin concentration (ng/g) in roots of OsNMCP1-OE-5, osnmcp1-1 mutants, and corresponding WT plants. Error bars represent the standard deviation (SD) of three replicates. Bar, 10 cm. Statistical significance was determined by T-test: **, P < 0.01.
Figure 17 illustrates the influence of OsNMCP1 on the growth of rice roots and the rate of cell division. (a) Phenotypic characterization and statistics of primary root length in two independent lines of ZH11 and OsNMCP1-OE plants; (b) Phenotypic characterization and statistics of primary root length in Dongjing (DJ) and Huayoung (HY) varieties, as well as osnmcp1-1 and osnmcp1-2 mutant lines; (c) Morphology of the root tip including MaZ (mature zone), EZ (elongation zone), MeZ (meristematic zone), and RCZ (root cap zone); (d) Expression levels of OsNMCP1 in different root tip zones analyzed by qRT-PCR; (e) Longitudinal view of Edu-labeled cells in the root meristematic tissue of 7-day-old seedlings of OsNMCP1-OE, osnmcp1 mutants, and corresponding wild-type (WT) plants; (f) Statistics of endogenous auxin concentration (ng/g) in roots of OsNMCP1-OE-5, osnmcp1-1 mutants, and corresponding WT plants. Error bars represent the standard deviation (SD) of three replicates. Bar, 10 cm. Statistical significance was determined by T-test: **, P < 0.01.
OsNMCP1 Regulates Root Growth and Drought Resistance by Altering Chromatin Accessibility
To investigate whether OsNMCP1 is involved in gene expression regulation associated with chromatin states, a combined analysis of ATAC-seq and RNA-seq was conducted on root tissues of OsNMCP1-OE-5 and ZH11 plants under normal and drought conditions. Compared to ZH11 under both conditions, a significant enrichment of sequencing reads near transcription start sites (TSS) was observed in OsNMCP1-OE lines (Figure 18a). Peak distribution analysis revealed that in OsNMCP1-OE lines, the upregulated differential accessibility regions (DARs) were predominantly concentrated around the TSS of adjacent genes, while the distribution of downregulated DARs showed no apparent pattern (Figure 18b), indicating that the increased chromatin accessibility mediated by OsNMCP1-OE primarily occurs in the TSS region. Additionally, the number of upregulated DARs was significantly higher than downregulated DARs under both normal (8791 vs 1524) and drought (12742 vs 2511) conditions (Figure 18c).
Furthermore, the genomic distribution characteristics of upregulated DARs exhibited a prominent enrichment pattern at promoters, with over 65% of peaks located within the promoter region (-3500 to +100 bp from TSS) (Figure 18c). These findings suggest that OsNMCP1 may predominantly act as a positive regulator of chromatin accessibility and modulate downstream gene expression by enhancing accessibility at their promoter regions.
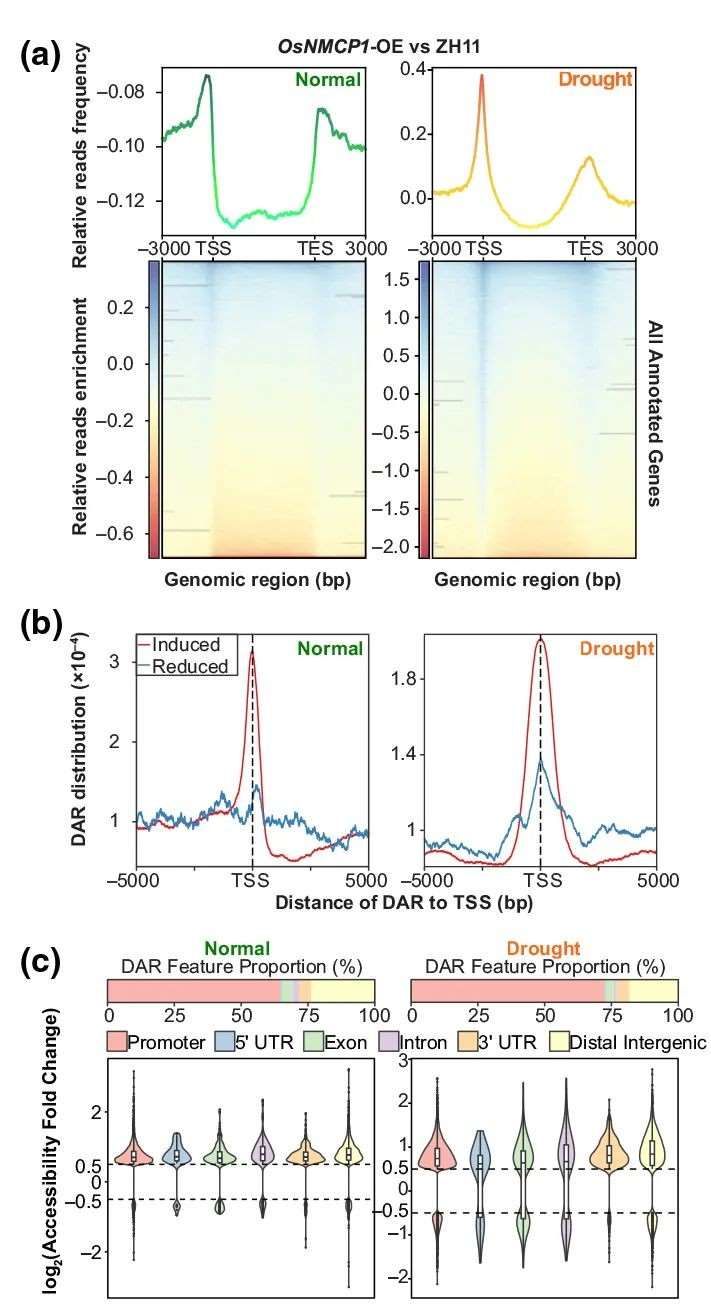 Figure 18 elucidates the alteration of chromatin accessibility in rice facilitated by OsNMCP1 overexpression. (a) Depicts the relative coverage curves of OsNMCP1-OE compared to ZH11 in ATAC-seq under normal and drought conditions. The line graph above illustrates the relative distribution frequency of OsNMCP1-OE normalized reads across annotated genomic regions. The heatmap below portrays the relative reads coverage within the -3000bp to +3000bp genomic regions of annotated genes. (b) Presents peak curve plots illustrating the distribution of upregulated or downregulated differentially accessible regions (DARs) in OsNMCP1-OE compared to ZH11 across genomic regions of all annotated genes. Red denotes upregulated DARs, while blue indicates downregulated DARs. (c) Represents the proportionate distribution of DARs across genomic regions. The bar plot above displays the distribution ratio of genomic regions for upregulated DARs in OsNMCP1-OE (considering only the features of the nearest genes to DARs). The violin plot below, with log2(Accessibility Fold Change), elucidates the abundance of DARs in each genomic region distribution.
Figure 18 elucidates the alteration of chromatin accessibility in rice facilitated by OsNMCP1 overexpression. (a) Depicts the relative coverage curves of OsNMCP1-OE compared to ZH11 in ATAC-seq under normal and drought conditions. The line graph above illustrates the relative distribution frequency of OsNMCP1-OE normalized reads across annotated genomic regions. The heatmap below portrays the relative reads coverage within the -3000bp to +3000bp genomic regions of annotated genes. (b) Presents peak curve plots illustrating the distribution of upregulated or downregulated differentially accessible regions (DARs) in OsNMCP1-OE compared to ZH11 across genomic regions of all annotated genes. Red denotes upregulated DARs, while blue indicates downregulated DARs. (c) Represents the proportionate distribution of DARs across genomic regions. The bar plot above displays the distribution ratio of genomic regions for upregulated DARs in OsNMCP1-OE (considering only the features of the nearest genes to DARs). The violin plot below, with log2(Accessibility Fold Change), elucidates the abundance of DARs in each genomic region distribution.
To substantiate this hypothesis, the same plant samples used for ATAC-seq underwent RNA-seq analysis to assess whether these chromatin changes were associated with alterations in adjacent gene expression. Integrated analysis revealed 362 and 619 common genes between differentially accessible genes (DAGs) and upregulated differentially expressed genes (DEGs) under normal and drought conditions, accounting for 49.93% and 20.35% of the total upregulated DEGs, respectively (Figure 19a). These genes were defined as Promoter-accessibility and Expression Increased Genes (PEIGs) under normal (N) and drought (D) conditions, namely NPEIGs and DPEIGs (Figure 19a). Gene Ontology (GO) enrichment analysis of PEIGs and DEGs indicated their association with stress response functions (Figure 19b), aligning with the role of OsNMCP1 in drought resistance.
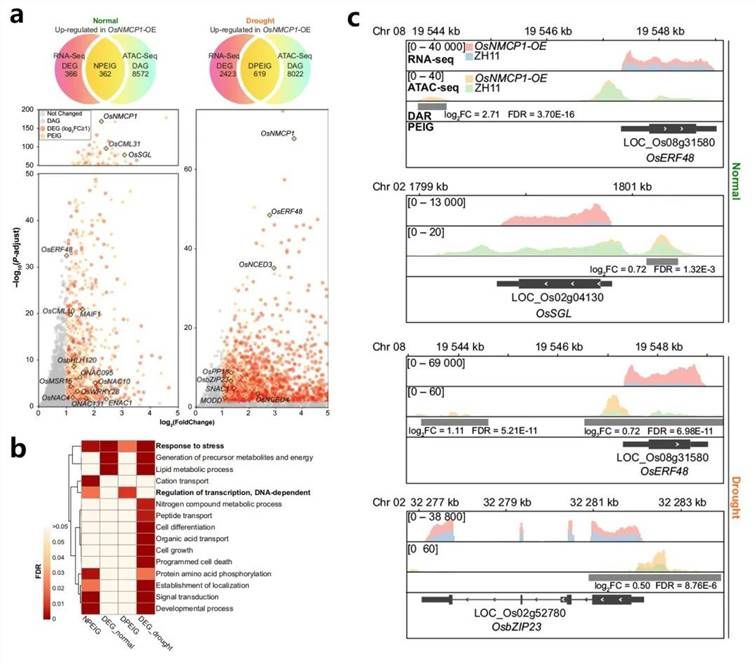 Figure 19 illustrates (a) the concurrence of genes upregulated in both ATAC-seq and RNA-seq datasets. The Venn diagram displays the overlap between genes upregulated in normal and drought conditions, termed NPEIGs and DPEIGs, respectively. The volcano plot, based on log2(Fold Change) and log10(q-value) of differentially expressed genes, delineates the distribution of PEIGs within the upregulated DEGs of OsNMCP1-OE strains under both conditions. (b) Presents the GO enrichment analysis of DEGs (excluding PEIGs), NPEIGs, and DPEIGs. The heatmap depicts the significance of GO term enrichment indicated by color intensity, with white representing non-significant GO terms. (c) Displays the standardized read density view of downstream regulatory genes of OsNMCP1, revealing alterations in regions of differential chromatin accessibility.
Figure 19 illustrates (a) the concurrence of genes upregulated in both ATAC-seq and RNA-seq datasets. The Venn diagram displays the overlap between genes upregulated in normal and drought conditions, termed NPEIGs and DPEIGs, respectively. The volcano plot, based on log2(Fold Change) and log10(q-value) of differentially expressed genes, delineates the distribution of PEIGs within the upregulated DEGs of OsNMCP1-OE strains under both conditions. (b) Presents the GO enrichment analysis of DEGs (excluding PEIGs), NPEIGs, and DPEIGs. The heatmap depicts the significance of GO term enrichment indicated by color intensity, with white representing non-significant GO terms. (c) Displays the standardized read density view of downstream regulatory genes of OsNMCP1, revealing alterations in regions of differential chromatin accessibility.
It is noteworthy that both PEIGs datasets are enriched in the "transcription regulation" Gene Ontology (GO) term, whereas DEGs are not enriched in this term. Further examination of the composition of PEIGs revealed that 17.4% of NPEIGs and 7.9% of DPEIGs encode transcription factors (TFs), constituting the largest category aside from unidentified proteins, including SNAC1 and OsbZIP23 (Figure 19a, c). These findings suggest that OsNMCP1 may regulate genes associated with drought response and root growth.
Interaction Between OsNMCP1 and the SWI/SNF Chromatin Remodeling Complex Component OsSWI3C
It has been previously reported that the chromatin remodeling factor AtSWI3B can interact with the nuclear matrix (Sarnowski et al., 2002). Given the influence of OsNMCP1 overexpression on chromatin accessibility, a yeast two-hybrid (Y2H) analysis was conducted to ascertain whether OsNMCP1 interacts with any homologous components of the chromatin remodeling complex (CRC) in rice, including OsSWI3A, OsSWI3B, OsSWI3C, OsSWI3D, and OsSWI3E. The results indicated that OsNMCP1 specifically interacts with OsSWI3C (LOC_Os11g08080) (Figure 20a).
To further validate the interaction between OsNMCP1 and OsSWI3C, bimolecular fluorescence complementation (BiFC) and co-immunoprecipitation (Co-IP) assays were performed using rice protoplasts. Both assays confirmed the direct interaction between OsNMCP1 and OsSWI3C in rice (Figure 20b, c).
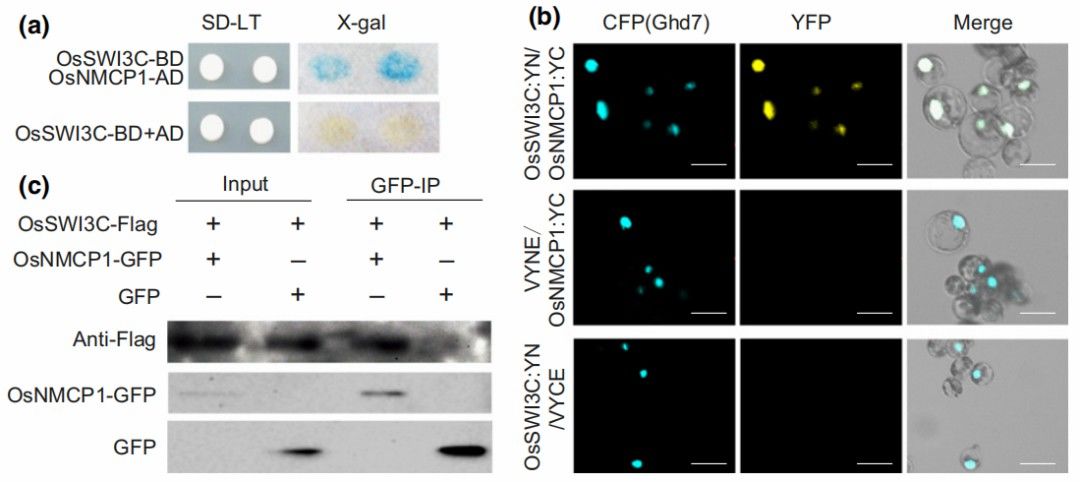 Figure 20 illustrates the interaction between OsNMCP1 and the SWI/SNF chromatin remodeling complex OsSWI3C. (a) Demonstrates the detection of their interaction via yeast two-hybrid assay. (b) Confirms the interaction in rice protoplasts using BiFC assay. Representative cellular images obtained through laser scanning confocal microscopy are provided. The detection of SWI3C
Figure 20 illustrates the interaction between OsNMCP1 and the SWI/SNF chromatin remodeling complex OsSWI3C. (a) Demonstrates the detection of their interaction via yeast two-hybrid assay. (b) Confirms the interaction in rice protoplasts using BiFC assay. Representative cellular images obtained through laser scanning confocal microscopy are provided. The detection of SWI3C
and NMCP1
interaction in rice protoplasts is denoted by yellow signals, with cyan signals representing the nuclear-localized protein Ghd7
. Scale bars indicate 10 μm. (c) Validates the interaction between OsNMCP1 and OsSWI3C in rice protoplasts through Co-IP assay.
OsSWI3C Negatively Regulates Drought Tolerance in Rice
To assess whether OsSWI3C also participates in drought response, transgenic lines overexpressing (3C-OE) and RNA interference (3C-RNAi) lines targeting OsSWI3C were generated, and their drought tolerance during the seedling stage was evaluated. Following recovery from drought stress, OsSWI3C-OE and OsSWI3C-RNAi lines exhibited significantly lower and higher survival rates, respectively, compared to the wild type ZH11, indicating a negative role of OsSWI3C in drought tolerance (Figure 21a-c). Immunofluorescence staining (IF) revealed the nuclear distribution of OsSWI3C under normal conditions and ABA treatment, with punctate patterns observed in the nucleoplasm but not in the nucleolus, suggesting its role as a nuclear protein and negative regulation under drought stress.
It has been reported that the chromatin remodeling complex (CRC) participates in transcriptional regulation by altering chromatin accessibility. Since OsSWI3C-RNAi plants exhibited enhanced drought tolerance, further investigation was conducted to determine whether the suppression of OsSWI3C affects chromatin accessibility of genes in OsNMCP1-OE. Comparative analysis of ATAC-seq results between OsSWI3C-RNAi and OsNMCP1-OE revealed a high similarity in the upregulated genes under drought stress, with strong enrichment around the TSS region (Figure 21d). However, under normal growth conditions, the distribution around the TSS region was significantly reduced in OsSWI3C-RNAi lines (Figure 21d). Comparison of the results between OsSWI3C-RNAi and OsNMCP1-OE lines showed 544 common genes (CoPEIGs), accounting for 87.88% of DPEIGs, indicating a shared regulation between OsNMCP1 and OsSWI3C (Figure 21e). Analysis of RNA-seq data extracted from the rice expression database revealed 884 and 726 co-expressed genes with correlation coefficients greater than 0.7 (Figure 21f), suggesting that OsNMCP1 and OsSWI3C may jointly regulate downstream gene expression through different pathways under drought stress.
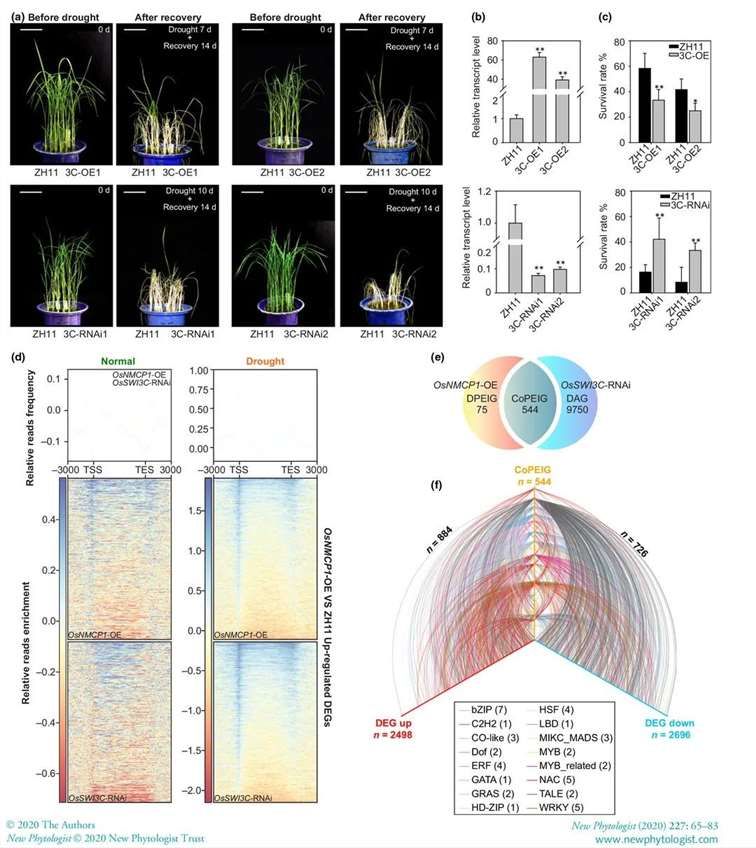 Figure 21: Impact of OsSWI3C on Rice Drought Tolerance and Chromatin Accessibility
Figure 21: Impact of OsSWI3C on Rice Drought Tolerance and Chromatin Accessibility
(a) Phenotypes of ZH11 vs. OsSWI3C-OE and ZH11 vs. OsSWI3C-RNAi plants before and after drought stress recovery during seedling stage. Each experiment's drought treatment and recovery time are indicated.
(b) qRT-PCR expression analysis of OsSWI3C in ZH11 and two independent OsSWI3C-OE lines or two independent OsSWI3C-RNAi lines.
(c) Survival rates of ZH11 and OsSWI3C-OE or OsSWI3C-RNAi plants after treatment in (a). 3C-OE: OsSWI3C overexpressing plants; 3C-RNAi: OsSWI3C RNA interference plants.
(d) ATAC-seq analysis of read coverage maps of OsNMCP1-OE or OsSWI3C-RNAi vs. ZH11 for upregulated differentially expressed genes (DEGs) under normal and drought conditions. The line plot above shows the relative distribution frequencies of OsNMCP1-OE and OsSWI3C-RNAi, normalized reads along the genomic regions of upregulated DEGs of OsNMCP1-OE. The heatmap below indicates the relative read coverage counts in the genomic regions of each upregulated DEG of OsNMCP1-OE.
(e) Venn diagram showing shared genes between drought-induced promoter accessibility upregulated differentially accessible genes (DAGs) in OsNMCP1-OE and DPEIGs in OsSWI3C-RNAi under drought conditions.
(f) Co-expression relationships of transcription factor (TF) genes in CoPEIG and OsNMCP1-OE plant DEGs under drought stress conditions. The orange, red, and blue axes represent CoPEIG, upregulated DEGs, and downregulated DEGs, respectively.
Conclusion:
OsNMCP1 localizes to the peripheral region of the nucleus and regulates chromatin accessibility.
OsSWI3C plays a crucial role in the gene expression regulation mediated by OsNMCP1.
Regulatory Mechanism:
The SWI/SNF chromatin remodeling complex subunit, OsSWI3C, is recruited to alter the structure of the nucleosome, inhibiting chromatin accessibility and resulting in the silencing of genes associated with root growth and drought tolerance. Drought stress induces the expression of OsNMCP1 protein. The elevated levels of OsNMCP1 protein may lead to more encounters between OsNMCP1 and OsSWI3C at the nuclear periphery, facilitating their interaction. This interaction may potentially release OsSWI3C molecules from the SWI/SNF complex, thereby increasing chromatin accessibility in genes associated with root growth and drought tolerance, leading to their upregulation.
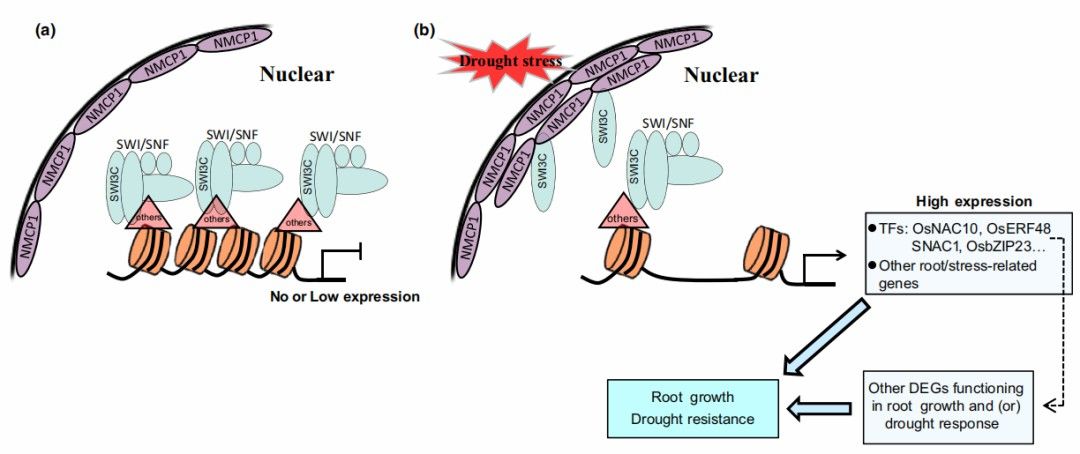 Figure 22 depicts a putative gene regulatory model of OsNMCP1 and OsSWI3C during root growth and drought response processes in rice (Oryza sativa).
Figure 22 depicts a putative gene regulatory model of OsNMCP1 and OsSWI3C during root growth and drought response processes in rice (Oryza sativa).
The two shared articles primarily focus on the application of ATAC-seq in mapping chromatin accessibility, identifying key transcription factors, recognizing promoter regions, and searching for transcription factor-regulated target genes. ATAC-seq, when combined with transcriptome sequencing, is best suited for mapping studies. Functional validation of crucial transcription factors and their regulated target genes, along with nucleic acid-protein interaction validation, enhances experimental data comprehensiveness and experimental design rigor.
References
Terms & Conditions Privacy Policy Copyright © CD Genomics. All rights reserved.